


What is Bupivacaine?
Sep 24,2020
History
Bupivacaine was synthesized simultaneously with mepivacainein 1957 but was at first overlooked because of the increasedtoxicity compared with mepivacaine. When themethyl on the cyclic amine of mepivacaine is exchanged fora butyl group the lipophilicity, potency and the duration ofaction all increase. Literature reports of cardiovascular toxicity,including severe hypotension and bradycardia, areabundant in the literature.91 Bupivacaine is highly bound toplasma proteins (95%), and thus the free concentration mayremain low until all of the protein binding sites are occupied.After that point, the plasma levels of bupivacaine rise rapidlyand patients may progress to overt cardiac toxicity withoutever showing signs of CNS toxicity. The cardiotoxicity ofbupivacaine is a result of its affinity to cardiac tissues and itsability to depress electrical conduction and predispose theheart to reentry types of arrhythmias. The cardiotoxicity ofbupivacaine was found to be significantly more prominentwith the “R” isomer, or the racemic mixture, thus the “S”stereoisomer is now on the market as levobupivacaine.
Local anesthetic
Bupivacaine is the most potent and toxic of all amide anesthetics. It is four times more potent than lidocaine, mepivacaine, and prilocaine, and three times more potent than articaine. It is four times more toxic than lidocaine, mepivacaine, and articaine, and six times more toxic than prilocaine. Pharmacologically, bupivacaine is structurally similar to mepivacaine, except the butyl group in bupivacaine is exchanged for the methyl group in mepivacaine. This substitution allows for a fourfold increase in potency, allows for toxicity, and provides bupivacaine with a major advantage of increased duration of action compared with other amides.4,6 Bupivacaine is the only anesthetic that provides a long duration of action, and does so despite its intense vasodilating properties, second only to procaine and significantly more than lidocaine, and is therefore only formulated with 1:200,000 epinephrine. Bupivacaine is highly lipid soluble, and binds powerfully to protein receptor sites in the sodium channels. It provides approximately 1.5 to 3 hours of pulpal anesthesia, and 4 to 9 hours of soft tissue anesthesia. Due to its long duration, bupivacaine may not be clinically practical for many dental procedures, including nonsurgical periodontal therapy. In an overdose, bupivacaine has equal effects on the CNS and cardiovascular system. Bupivacaine’s long half-life (2.7 hours) further increases the risk for systemic toxicity.
Usage & Metabolism
The use of bupivacaine is indicated when long pulpal anesthesia is needed (greater than 1.5 hours) and/or postoperative pain control is expected to be needed (e.g., periodontal surgery, endodontics, oral surgery). In many situations, it may be prudent to administer after long procedures have been completed and immediately before the patient’s discharge from the office. This provides the benefit of extended pain control, allowing time for oral pain medications to take effect. In addition, it is a good alternative when profound anesthesia has been difficult to attain with other anesthetic formulations. With the highest pKa of the amide anesthetics, bupivacaine has a slightly slower onset of action, but the duration of anesthesia is almost twice that of lidocaine. Bupivacaine is not recommended for use on patients who are prone to self-mutilation (patients with special needs and young children). It is metabolized by liver enzymes in the same manner as lidocaine and mepivacaine, and up to 16% is excreted unchanged by the kidneys. Individuals with significant hepatic disease can receive bupivacaine but at a reduced dose. Table 5-23 lists the main properties of bupivacaine, and Table 5-24 gives helpful tips for anesthetic selection and precautions for bupivacaine.
The MRD for bupivacaine is 0.9 mg/lb or 2.0 mg/kg (based on Canadian weight-based recommendations, there is no FDA weight-based recommendations), and the absolute MRD is 90 mg.
Manufacturing Process
121 parts by weight of 2.6-xylidine are heated with 400 parts of diethylmalonate at 160°C for 1 hour, and the alcohol formed by the reaction is allowed to distill off. Thereafter the reaction mass is cooled to 80°C, and 500 parts of alcohol are added. After cooling the dixylidide is sucked off, and the alcohol solution with malonic ester monoxylidide is poured into 2,000 parts of water. The monoxylidide precipitates, is filtered off and washed with water, and recrystallized in diluted alcohol. Nitrosation thereafter takes place by dissolving the dried monoxylidide in chloroform and by introducing nitrosyl chloride at 0°C until the nitrosation is completed. The isonitrosomalonic ester xylidide is filtered off and dried. Thereafter the reduction takes place with zinc powder and formic acid at 90°-100°C.
The formic acid is distilled off, and the remainder dissolved in warm benzene and washed with a bicarbonate solution to a neutral reaction. After the benzene has been distilled off, the aminomalonic ester xylidide is obtained. This is treated with an equal quantity of sodium ethylate and boiled with twice the theoretical quantity of tetramethylene bromide in absolute alcohol. After 6 hours of boiling, the sodium bromide formed is separated, and the mixture is steamdistilled in order to remove the excess of tetramethylene bromide. The remaining oil, which mainly consists of deltabromobutylaminomalonic ester xylidide is separated from the water and boiled with 3 parts of concentrated hydrochloric acid for 3 hours. Thereafter carbonfiltering and evaporation to dryness under vacuum takes place. The residue is dissolved in water, and the pH adjusted with sodium hydroxide to 5.5. The solution is extracted twice with ether, and the water is made strongly alkaline with sodium hydroxide.
The oil precipitates and is crystallized after a time. The crystals are separated and dried under vacuum. The pipecolyl-2,6-xylidide produced is alkylated by boiling for 10-20 hours with 0.6 part n-butylbromide in an n-butanol solution in the presence of 0.5 part potassium carbonate. The potassium carbonate is filtered off and the butanol is distilled off in vacuum. The residue is dissolved in diluted hydrochloric acid and carbon treated, after which the base is precipitated with sodium hydroxide in the form of white crystals, which are filtered off and washed with water. The base obtained, which consists of N-n-butyl-pipecolyl-2,6-xylidide is sufficiently pure for the production of salts.
- Related articles
- Related Qustion
- Liposomal bupivacaine: Overview, Efficacy in Postoperative Pain and Safety Mar 6, 2024
Liposomal bupivacaine provides extended postoperative pain relief with good safety. Adverse effects include nausea, vomiting, and headache. Caution is advised for wider application.
- Bupivacaine: mechanism of action, clinical applications and safety Nov 13, 2023
Bupivacaine is a local anesthetic that blocks nerve impulses, providing temporary numbness in specific areas of the body.
Ferric nitrate nonahydrate compound is also known as ferric nitrate, Fe(NO3)3·9H2O=404.02, light purple or off-white monoclinic crystal, easy to deliquesce.....
Sep 23,2020Inorganic saltsLeuprorelin/Leuprolideacetate (LA) is a highly active analogue (LHRH-A, GnRHA) of luteinizing hormone releasing hormone (LHRH, or gonadotropin releasing hormone, GnRH) produced by the hypothalamus.....
Sep 25,2020API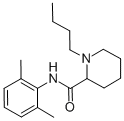
Bupivacaine
2180-92-9You may like
- Bupivacaine
-

- $0.00 / 1KG
- 2024-04-24
- CAS:2180-92-9
- Min. Order: 1KG
- Purity: 99%
- Supply Ability: 500
- Bupivacaine
-

- $3.00 / 1kg
- 2024-04-12
- CAS:2180-92-9
- Min. Order: 1kg
- Purity: 99.9%
- Supply Ability: 10 tons
- Bupivacaine
-

- $0.00 / 1KG
- 2024-03-16
- CAS:2180-92-9
- Min. Order: 100g
- Purity: 98%+
- Supply Ability: 100kg