- Acrivastine
-

- $31.00 / 5mg
-
2024-10-31
- CAS:87848-99-5
- Min. Order:
- Purity: 99.85%
- Supply Ability: 10g
- Acrivastine
-

- $1.00 / 1g
-
2021-07-20
- CAS:87848-99-5
- Min. Order: 1g
- Purity: 99%
- Supply Ability: 50tons
- Acrivastine
-

- $15.00 / 1KG
-
2021-07-13
- CAS:87848-99-5
- Min. Order: 1KG
- Purity: 99%+ HPLC
- Supply Ability: Monthly supply of 1 ton
|
| | Acrivastine Basic information |
| Product Name: | Acrivastine | | Synonyms: | (e)-3-[6-[(e)-1-(4-methylphenyl)-3-pyrrolidin-1-ylprop-1-enyl]pyridin-2-yl]prop-2-enoic acid;ACRIVASTINE;Acrivastin;(E,E)-3-[6-[1-(4-Methylphenyl)-3-(1-pyrrolidinyl)-1-propenyl]-2-pyridinyl]-2-propenoic acid;BW-270C;BW-825C;BW-A825C;(E)-3-[6-[(E)-1-(4-Methylphenyl)-3-(1-pyrrolidinyl)-1-propenyl]-2-pyridinyl]propenoic acid | | CAS: | 87848-99-5 | | MF: | C22H24N2O2 | | MW: | 348.44 | | EINECS: | | | Product Categories: | API | | Mol File: | 87848-99-5.mol | 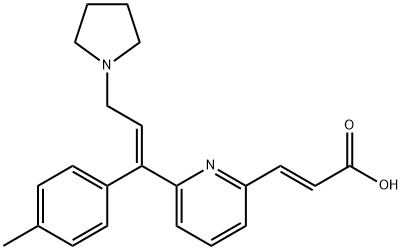 |
| | Acrivastine Chemical Properties |
| Melting point | 222° (dec) | | Boiling point | 555.1±50.0 °C(Predicted) | | density | 1.170±0.06 g/cm3(Predicted) | | storage temp. | 2-8°C | | solubility | DMSO: >2mg/mL (warmed) | | form | powder | | pka | 1.99±0.10(Predicted) | | color | white to beige | | Stability: | Light Sensitive | | CAS DataBase Reference | 87848-99-5(CAS DataBase Reference) |
| WGK Germany | 3 | | RTECS | UD3474000 |
| | Acrivastine Usage And Synthesis |
| Description | Acrivastine is an orally-active HI .receptor antagonist reportedly useful in the treatment of
allergic rhinitis. The main advantages of acrivastine in comparison to older antihistamines
are its low sedative potential and the absence of anticholinergic side-effects. | | Description | Acrivastine is a histamine H1 receptor antagonist with a Ki value of 10 nM in COS-7 cells expressing the human receptor. In vivo, acrivastine (1 mg/kg) completely inhibits response to histamine in guinea pigs. Formulations containing acrivastine have been used for the treatment of seasonal allergies and hay fever. | | Originator | Burroughs Wellcome (USA) | | Uses | Antihistaminic. | | Uses | Acrivastine has been used as an antihistamine to investigate the relation between the increased residence time of antihistamine at the histamine H1 receptor (H1R) and the duration of effective target-inhibition by this antagonist. | | Definition | ChEBI: A member of the class of pyridines that is (pyridin-2-yl)acrylic acid substituted at position 6 by a [(1E)-1-(4-methylphenyl)-3-(pyrrolidin-1-yl)prop-1-en-1-yl group. It is a non-sedating antihistamine used for treatment of hayfever, urtic
ria, and rhinitis. | | Manufacturing Process | Butyl lithium (50 ml, 1.65 mol in hexane) was added under nitrogen to a
stirred suspension of 2,6-dibromopyridine (19.5 g) in dry ether (200 ml) at -
50°C. After 0.75 h a solution of 4-tolunitrile (10.0 g) in ether (50 ml) was
added; stirring was continued at -50°C for 3 h. The mixture was allowed to
warm to -30°C and treated with hydrochloric acid (200 ml, 2 mol). The
precipitated solid was collected, washed with water to give the 2-bromo-6-(4-
toluoyl)pyridine as colourless needles (12.2 g), melting point 97°-98°C
(recrystallized from aqueous ethanol).
A mixture of 2-bromo-6-(4-toluoyl)pyridine (200.0 g), ethylene glycol (85 ml),
p-toluenesulphonic acid (32.0 g) and benzene (11 ml) was boiled under a
Dean/Stark trap until water collection had become very slow (about 20 ml
collected in 16 h).
The cooled solution was poured into ice/water containing sodium carbonate
(100.0 g) with stirring. The benzene layer was separated, washed with water,
dried with sodium sulfate and evaporated to about 500 ml. Cooling gave a first crop of 2-(6-bromo-2-pyridyl)-2-(4-tolyl)-1,3-dioxolan, melting point
113°-114°C (170.0 g). Dilution with petroleum ether gave a second crop,
melting point 109°-112°C (34.0 g). The residue after evaporation (31.0 g)
was recycled.
A solution of 2-(6-bromo-2-pyridyl)-2-(4-tolyl)-1,3-dioxolan, vide supra, (70.0
g) in dry toluene (800 ml) was added dropwise during 5 h to a stirred solution
of butyl lithium (1.6 mol in hexane, 200 ml) and toluene (200 ml) at -65° to -
72°C under nitrogen. After a further 30 min at -70°C, dry dimethylformamide
(40 ml) was added during 35 min. Stirring continued overnight at -70° to -
60°C. Hydrochloric acid (2 N, 400 ml) was added, allowing the temperature to
rise to about -10°C. After 30 min, 2 N ammonia (ca. 90 ml) was added to pH
7-8. The toluene layer was separated and the aqueous phase was extracted
with ether. The combined organic liquids were washed with ice/water, dried
(MgSO4) and evaporated in vacuum below 50°C. The aldehyde, 2-(6-formyl-2-
pyridyl)-2-(4-tolyl)-1,3-dioxolan, (63.9 g) crystallized on keeping at 3°C,
melting point 52-63°C.
The 2-(6-formyl-2-pyridyl)-2-(4-tolyl)-1,3-dioxolan (2.5 g) was dissolved in
1,2-dimethoxyethane (10 ml) and added to a solution of the phosphonate
carbanion produced from triethyl phosphonoacetate (2.0 g) and sodium
hydride (0.22 g) in the same solvent. The mixture was stirred for 2 h, diluted
with ether (25 ml) and treated with hydrochloric acid (5 ml, 2 mol). The
organic phase was separated, washed with water, dried, and evaporated. The
resulting oil was dissolved in ethanol (20 ml) containing concentrated
hydrochloric acid (3 ml) and water (3 ml). After heating on the steam bath for
10 min, the solution was diluted with ice water, rendered alkaline with sodium
bicarbonate solution, and extracted with ether. Evaporation gave 1.0 g ((E)-3-
(6-(4-toluoyl)-2-pyridyl)acrylate as colourless platelets, melting point 108°-
111°C (crystallized from cyclohexane).
Butyl lithium (10 ml, 1.64 mol in hexane) was added under nitrogen to a
stirred suspension of triphenyl-2-pyrrolidinoethylphosphonium bromide (7.2 g)
in dry toluene (75 ml). After 0.5 h, ((E)-3-(6-(4-toluoyl)-2-pyridyl)acrylate,
vide supra, (4.8 g) in toluene (50 ml) was added. The suspension, initially
orange, became deep purple, then slowly faded to yellow during 2 h heating
at 75°C. The cooled solution was diluted with ether (150 ml) and treated with
hydrochloric acid (50 ml, 2 mol). The aqueous phase was separated, washed
with ether, and basified with potassium carbonate (ice) and extracted with
ether. The mixture of isomeric esters obtained by evaporation was dissolved in
ethanol (100 ml) containing sodium hydroxide solution (20 ml, 1 mol) and
partially evaporated on the steam bath under reduced pressure for 5 min. The
residual aqueous solution was neutralized with sulfuric acid (20 ml, 0.5 mol)
and evaporated to dryness. The solid residue was extracted with hot
isopropanol (3x50 ml) and the extracts were concentrated until crystallization
commenced. The (E)-3-(6-(3-pyrrolidino-1-(4-tolyl)prop-1-(E)-enyl)-2-
pyridyl)acrylic acid, melting point 222°C (dec. recrystallization from
isopropanol) was obtained. | | Brand name | Semprex | | Therapeutic Function | Antihistaminic | | General Description | Acrivastine, (E, E)-3-[6-[1-(4-methylphenyl)-3-(1-pyrrolidinyl)-1-propenyl-2-pyridinyl]-2-propenoic acid(Semprex), is a fixed-combination product of the antihistamineacrivastine (8 mg) with the decongestant pseudoephedrine(60 mg). Acrivastine is an odorless, white to pale cream crystallinepowder that is soluble in chloroform and alcohol andslightly soluble in water.
Acrivastine is an analog of triprolidine containing acarboxyethenyl moiety at the 6-position of the pyridylring. Acrivastine shows antihistaminic potency and durationof action comparable to those of triprolidine (Table23.2). Unlike triprolidine, acrivastine does not display significantanticholinergic activity at therapeutic concentrations.Also, the enhanced polarity of this compound resultingfrom carboxyethenyl substitution limits BBBaccumulation, and thus, this compound produces less sedationthan triprolidine.
Limited pharmacokinetic data are available for this compound.Orally administered drug has a half-life of about 1.7hours and a total body clearance of 4.4 mL/min per kilogram.The mean peak plasma concentrations are reported tovary widely, and the drug appears to penetrate the CNSpoorly. The metabolic fate of acrivastine has not beenreported. | | Biochem/physiol Actions | Acrivastine is a second-generation antihistamine, an H1-receptor antagonist. | | Clinical Use | Acrivastine, an acidic congener of triprolidine in which a carboxylic acid–substituted chain
has been attached, also is a second-generation, nonsedating antihistamine. Penetration of
the blood-brain barrier is limited, and it is less sedating than triprolidine. It is used principally
in a combination with a decongestant. | | Drug interactions | Potentially hazardous interactions with other drugs
Antivirals: concentration possibly increased by
ritonavir. | | Metabolism | Acrivastine undergoes metabolism in the liver, and along
with an active metabolite, is excreted principally in the
urine. |
| | Acrivastine Preparation Products And Raw materials |
|