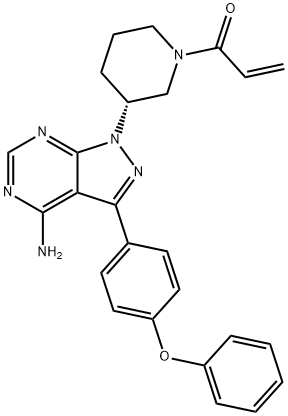
Ibrutinib: A Narrative Drug Review
Apr 7,2023
General Description
Ibrutinib (PCI-32765) is a novel, orally active, irreversible inhibitor of BTK that has demonstrated considerable efficacy in a variety of B-cell malignancies. The US Food and Drug Administration (FDA) formally designated ibrutinib as a breakthrough therapy, and it was approved for the treatment of relapsed and refractory MCL on November 2013 and for relapsed and refractory CLL on February 2014.7–10 Breakthrough therapy designation is meant to expedite the development and review of drugs that are intended to treat a serious condition and have sufficient preliminary clinical evidence that indicates that the drug may demonstrate substantial improvement in at least one clinically significant endpoint over currently available therapies. Other anticancer drugs that received breakthrough therapy designation included obinutuzumab in 2013 and, ofatumumab, ceritinib, and idelalisib in 2014.1

Figure 1. Properties of Ibrutinib
Mechanism of Action
In the B-cell receptor (BCR) signaling pathway, after an antigen triggers the BCR, tyrosine protein kinases Lyn and Syk activate BTK.2 BTK then Activates phosphatidylinositol 3 kinase (PI3K), which converts phosphatidylinositol (4,5)-bisphosphate (PIP2) to phosphatidylinositol (3,4,5)-trisphosphate (PIP3). This leads to an increase in intracellular calcium release, activation of AKT (Protein Kinase B), and downstream activation of the mammalian target of rapamycin (mTOR) pathway. In addition, BTK activates 1-phosphatidylinositol-4,5-bisphosphate phosphodiesterase gamma-2 (PLCg2), which promotes the release of intracellular calcium, diacylglycerol (DAG), and inositol triphosphate (IP3). This leads to an increase in nuclear factor kappa B (NF-kB), thereby inhibiting apoptosis of the B-cell and promoting cancer. Ibrutinib is a small-molecule inhibitor of BTK that binds covalently to Cys481 on the BTK active site, leading to an irreversible inhibition at Tyr223. BTK inhibition by ibrutinib prevents malignant B-cell substrate adhesion, migration, proliferation, and survival.2
Therapeutic Uses
Advani et al. conducted a phase 1, dose-finding study evaluating the use ibrutinib in 56 patients (median age, 65 years; range, 41 to 82) with relapsed or refractory CLL/SLL, DLBCL, FL, MCL, marginal zone lymphoma (MZL), or Waldenstrom macroglobulinemia (WM) who failed at least one prior therapy. Of the 50 patients who were evaluable for tumor response, 60% achieved an objective response, with 54% achieving an overall response rate (ORR) in the intention-to-treat (ITT) population. Responses were seen in patients with all histologies, with the highest response rates seen in MCL (79%) and CLL/SLL (69%). The median progression-free survival (PFS) was 13.6 months and 20 patients remained on the study at the time of data cutoff.16 The results of this study then led to further investigation of ibrutinib.3
Pharmacokinetics
Ibrutinib is rapidly absorbed after oral administration with a Tmax of 1–2 h. Its Cmax is 35 ng/ml, and the area under the curve (AUC) at a dose of 560 mg is 953 ± 705 ng.h/ml and at a dose of 420 mg is 680 ± 517 ng.h/mL. Its action begins 4 h after oral administration and is sustained for at least 24 h, thus explaining the rationale for once-a-day administration. Although food increases its exposure compared to the fasting state, there are no recommended food restrictions. In plasma, it binds irreversibly to albumin and alpha-glycoprotein. Protein binding can account for 97.3% of the administered dose. Ibrutinib is primarily metabolized by CYP3A5 and CYP3A4 and to a minor extent by CYP2D6. The major route of elimination is through the feces (80%). Less than 10% of the drug is excreted in the urine. The elimination half-life of ibrutinib is approximately 4–6 h.4
Adverse effects
Though ibrutinib is considered to be relatively well tolerated than conventional chemo-immunotherapy, it is associated with potentially serious side effects. Proper anticipation, monitoring, and appropriate management including dose modification are key to the optimal outcomes, as ibrutinib dose reduction has an adverse impact on disease control. Long-term follow-up studies with ibrutinib have revealed that up to 50% of the patients have discontinued the therapy, and the most common reason was disease progression. Roughly less than half of the dose discontinuations are because of toxicities, and, hence, appropriate patient selection and monitoring is required to obtain optimal outcomes. Most of the side effects of ibrutinib are mild (Grade 1/2) toxicities and are easily manageable. However, Grade 3/4 toxicities require either discontinuation of the drug or dose modification. Besides, lymphocytosis is seen in nearly two-thirds of the patients within the first month of ibrutinib therapy. This is accompanied by reduction in lymphadenopathy, thereby justifying it as a redistribution phenomenon without any clinical significance. It is reversible in 95% of the patients, and even prolonged lymphocytosis has not been shown to be predictive of relapse.5
Drug-drug interactions
Ibrutinib is a major substrate of CYP3A and a minor substrate of 2D6. Therefore, co-administration with moderate/strong inhibitors or inducers of these enzymes may potentially lead to clinically significant drug–drug interactions.6 In a phase 1 study, ibrutinib 120 mg was given on day 1 and 40 mg on day 7, along with a strong CYP3A inhibitor, ketoconazole 400 mg on days 4 to 9.6, ketoconazole increased the maximum plasma concentration (Cmax) of ibrutinib by 29-fold and AUC by 24-fold. Moderate CYP3A inhibitors, such as diltiazem and erythromycin may increase the AUC by 6- to 9-fold when ibrutinib is taken in a fasted state.6 In contrast, strong CYP3A inducers may decrease the Cmax and AUC by more than 10-fold, while moderate CYP3A inducers may decrease the AUC up to three-fold. The dose of ibrutinib should be reduced to 140 mg when given concomitantly with a moderate CYP3A inhibitor.6
Ibrutinib and its active metabolite (PCI-45227) are unlikely to inhibit CYP isoforms and both of these compounds are weak inducers of CYP in vitro. Although ibrutinib is not a P-glycoprotein (P-gp) substrate, it may lead to clinically significant effects on other P-gp substrates in the gastrointestinal tract. Specifically, the plasma levels of drugs with narrow therapeutic indices may be increased when ibrutinib is co-administered. Ibrutinib has also been implicated in affecting platelet aggregation in animal studies. Concomitant use of anticoagulants, such as warfarin and antiplatelet drugs, including aspirin, clopidogrel, prasugrel, should be used with caution due to the potential risk of bleeding.6
Molecular Mechanisms of Resistance
Resistance to ibrutinib and its underlying mechanisms have quickly become apparent as the drug is being used more widely. This has led to the exploration of potential combinations with other compounds in such refractory populations. Depending on the disease, varying proportions of patients show primary resistance. For example, 10%–30% of patients with CLL show no initial response to ibrutinib. Certain risk factors such as del(17p), p53 mutation, and complex karyotype have been reported. Similarly, DLBCL of germinal center origin, overexpression of CD79b, and CARD11 mutations have been linked to primary resistance in DLBCL. Overall, tumors with less active BCR signaling or mutations downstream to BTK in the BCR signaling pathway tend to be primarily refractory.7
A small proportion of patients have also been reported to have a gain-of-function mutation in PLCG2, which is downstream to BTK. In such cases, BTK inhibitors have no activity, but it opens a possibility for the inhibition of the SYK and LYN pathways. Deletion of 8p has also been reported in cases of secondary ibrutinib resistance. The deletion leads to haplo-insufficiency of the TNF-related apoptosis-inducing ligand receptors (TRAIL-R), thus leading to insensitivity to TRAIL.[88] Gain of chromosome 2p leads to the overexpression of exportin-1 gene (XPO1), which can potentially be targeted with selinexor. The upregulation of other cell signaling pathways such as the AKT, ERG, and noncanonical NF-KB pathways has also been reported with ibrutinib therapy and provides a rationale for combination therapy.7
Synthesis of Ibrutinib

Figure 2. Synthesis of Ibrutinib
The small-scale route starts from 4-phenoxybenzoyl chloride (1), which is reacted with malononitrile 2 to afford alkenol 3. This is methylated upon the addition of trimethylsilyldiazomethane to yield derivative 4. The addition of hydrazine to 4 yielded the amino-pyrazole 5, which is, in turn, converted to pyrazolo-pyrimidine 6 via adding formamide at 180 ◦C. The subsequent Mitsunobu reaction between 6 and (S)-tert-butyl 3-hydroxypiperidine-1-carboxylate 7 afforded derivate 8 with a corresponding inversion of configuration at the alcoholic carbon center. The BOC-removal from 8 gave the free piperidine derivative 9. This was acylated with acryloyl chloride to provide ibrutinib (10, Figure 2). This pathway allowed chemists to explore different heterocycle via Mitsunobu and different substituents at the free N-piperidine moiety.8
References
1. FDA approves Imbruvica to treat chronic lymphocytic leukemia. US Food and Drug Administration
2. Novero A, Ravella PM, Chen Y, et al. Ibrutinib for B cell malignancies. Exp Hematol Oncol 2014; 3: 4.
3. Advani RH, Buggy JJ, Sharman JP, et al. Bruton tyrosine kinase inhibitor ibrutinib (PCI-32765) has significant activity in patients with relapsed/refractory B-cell malignancies. J Clin Oncol 2013; 31: 88–94.
4. Advani RH, Buggy JJ, Sharman JP, et al. Bruton tyrosine kinase inhibitor ibrutinib (PCI-32765) has significant activity in patients with relapsed/refractory B-cell malignancies. J Clin Oncol 2013; 31: 88–94.
5. Deeks ED. Ibrutinib: A Review in Chronic Lymphocytic Leukaemia Drugs. 2017;77:225–36.
6. Product Information: Imbruvica (ibrutinib) oral capsules. Sunnyvale, CA: Pharmacyclics Inc, Feb 2014.
7. Woyach JA, Furman RR, Liu TM, Ozer HG, Zapatka M, Ruppert AS, et al Resistance mechanisms for the Bruton's tyrosine kinase inhibitor ibrutinib N Engl J Med. 2014;370:2286–94.
8. Benedetto Tiz, Davide, et al. "Top Selling (2026) Small Molecule Orphan Drugs: A Journey into Their Chemistry." International Journal of Molecular Sciences 24.2 (2023): 930.
- Related articles
- Related Qustion
- Ibrutinib: Uses, Mechanism of action and Side effects Apr 2, 2024
Ibrutinib is an oral, potent, selective and irreversible small molecule inhibitor of Btk. It is a prescription drug with the trade name IMBRUVICA?.
- Side effects of Ibrutinib Nov 4, 2021
Ibrutinib is an inhibitor of BTK kinase, which can covalently bind to the cysteine residues in the active center of BTK, thereby inhibiting the activity of B cells.
- What is Ibrutinib? Feb 10, 2020
Ibrutinib is a first-in-class,potent, orally administered covalently-binding inhibitor of BTK.
L(-)-Carvone is a member of a family of chemicals called terpenoids and exhibits multiple pharmacological properties such as antibacterial, antifungal, antiparasitic, antineuraminidase activities.....
Apr 7,2023APICrizotinib (XALKORI) is a potent and selective mesenchymal epithelial factor/anaplastic lymphoma kinase (c-Met/ALK) inhibitor.....
Apr 7,2023API