背景及概述
乙肝病毒感染已经成为全球性的危害。慢性乙肝可导致肝硬化、肝癌及肝功能衰竭,我国有数以亿计的乙肝患者和病毒携带者,全球每年因乙肝相关疾病的死亡人数高达百万之多。恩替卡韦是百时美施贵宝公司( Bristol -Myers Squib, BMS) 研究开发的抗乙肝病毒药物,2005年 4月首次获得美国 FDA 的批准。其化学名为 2-氨基-1, 9-二氢-9- [ (1S, 3R, 4S) -4-羟基-3-羟甲基-2-亚甲基环戊基] - 6H-嘌呤-6-酮一水合物。临床主要用于治疗成人伴有病毒复制活跃和血清转氨酶持续增高、或肝组织学为活动性病变的慢性乙型肝炎。
制备
恩替卡韦的合成路线主要有 2 条,条:以 ( 1S-反式) -2- [ (苯甲氧基) 甲基] -3-环戊烯-1-醇 (2) 为原料,经 8 步反应得到恩替卡韦,第二条:以 [ 1S- ( 1α, 2α, 3β,5α)] -2- [ ( 苯甲氧基) 甲基] -6-氧杂二环 [ 3, 1, 0] 己-3-醇为原料, 经 10 步反应, 或以 ( 3as, 6aR) -3, 3a,6, 6a-四氢-2H-环戊二烯并 [ b] 呋喃-2-酮为原料, 经 11 步反应制得目标化合物。2 种方法的主要区别在于前者是先在环戊基上引入嘌呤再进行亚甲基化, 而后者是先在环戊基上引入亚甲基再引入嘌呤。文献的反应步骤相对较长、工艺复杂,而且涉及方法专利。因此,本文以 (1S-反式) -2- [ ( 苯甲氧基) 甲基] -3-环戊烯-1-醇 ( 2) 为原料,经环氧化、保护羟基、定向加成、保护氨基、氧化、亚甲基化和两步脱保护基制得恩替卡韦[1]。其合成路线图如下图:
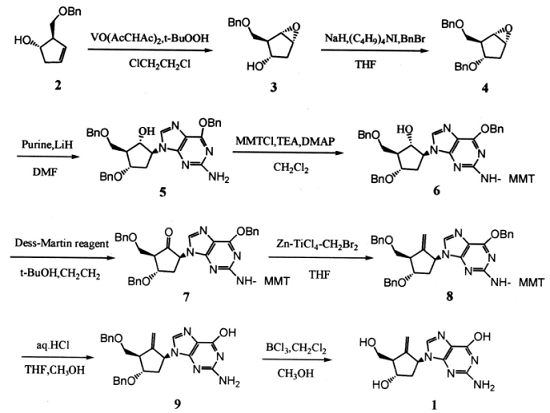
图1 恩替卡韦合成路线图
[ 1S- (1α, 2α, 3β, 5α)] -2- [ ( 苯甲氧基) 甲基] -6-氧杂二环 [ 3, 1, 0] 己-3-醇 (3) 的制备
过氧叔丁醇 90.1 g ( 1.0 mol) 和二氯甲烷 (100 mL)混合液滴加到 ( 1S-反式) -2- [ ( 苯甲氧基) 甲基] -3-环戊烯-1-醇 (2) 102.0 g (0.5 mol) , 乙酰丙酮氧钒 2.7 g (0.01mol) 和二氯甲烷 (200 mL) 的混合溶液中, 氮气保护, 室温搅拌反应 16 h, 降温至 0 ℃, 加入饱和亚硫酸钠溶液, 室温再搅拌 2 h, 分出有机层, 水层用二氯甲烷萃取, 合并有机相, 饱和食盐水洗涤, 无水硫酸钠干燥, 真空浓缩, 得( 3) 101.5 g 。 收率 92.3%。
[ 1S- (1α, 2α, 3β, 5α)] -3- (苯甲氧基) -2- [ (苯甲氧基) 甲基] -6-氧杂二环 [ 3, 1, 0] 己烷 (4) 的制备
氮气保护, 在干燥的三颈瓶中加入干燥 的四氢呋喃( 500 mL) 和氢化钠 ( 60%) 20.0 g (0.5 mol), 20℃以下滴加 (3) 99 g ( 0.45 mol) 的四氢呋喃 (100 mL) 溶液, 滴完后室温反应 2 h, 再向体系中加入四丁基碘化铵 1.8 g (5mmol) , 滴加溴苄 85.5 g ( 0.5 mol) , 室温搅拌反应过夜,加入乙醇破坏氢钠, 浓缩, 残留物中加入水 ( 350 mL) 和乙醚 (350 mL) , 分出有机相, 水层用乙醚 ( 100 mL ×2)萃取。 合并有机相, 无水硫酸镁干燥, 浓缩, 柱层析纯化梯度洗脱:石油醚-乙酸乙酯 =10∶1 洗至产物, 石油醚-乙酸乙酯 =3∶1 洗脱完产物。得 ( 4) 107.2 g, 收率 76.8%。
[ 1S- ( 1α, 2β, 3α, 5β)] -5- [ 2-氨 基-6- (苯甲氧基) -9H-嘌呤-9-基] -3- (苯甲氧基) -2- [ (苯甲氧基) 甲基]环戊醇 (5) 的制备
氮气保护下, 于干燥的三颈瓶中加入 2-氨基-6- (苯甲氧基) -9H-嘌呤 144.6 g (0.6 mol) 和干燥的 DMF ( 450 mL) ,搅拌溶解, 室温下加入氢化锂 2.52 g (0.315 mol) , 保持 10min, 升温至60 ℃反应 15 min, 60 ℃下滴加 (4) 93.0 g (0.3mol) 的 DMF (250 mL) 溶液。 60 ℃反应 15 min, 升温至125 ℃反应 3 h, 反应混合物降至室温, 加入冰醋酸 ( 20mL), 室温下搅拌 30 min, 反应混合物倒入水 ( 3 000 mL)中, 乙酸乙酯 (500 mL×4) 萃取, 合并有机相, 依次用水( 500 mL), 食盐水 ( 500 mL) 洗涤, 干燥, 真空浓缩, 加入二氯甲烷 (2 000 mL), 析出的固体 (未反应的 2-氨基-6-( 苯甲氧基) -9H-嘌呤) 过滤, 滤液真空浓缩, 残留物柱层析(洗脱剂:二氯甲烷-甲醇 =60∶1) , 得 (5) 98.5 g, 收率 59.6%。
[ 1S- ( 1α, 2β, 3α, 5β)] -5- [ 2- [ [ ( 4-甲 氧基苯基) -二苯甲基] 氨基] -6- ( 苯甲氧基) -9H-嘌呤-9-基] -3-( 苯甲氧基) -2- [ (苯甲氧基) 甲基]环戊醇 (6) 的制备
氮气保护, 室温下将 (5) 82.7 g ( 0.15 mol) 溶于二氯甲烷 (600 mL) 中, 分别向溶液中加入三乙胺 30.3 g ( 0.3mol) , 对甲氧基苯基二苯基氯甲烷 55.6 g ( 0.18 mol) 和 4-二甲氨基吡啶 1.2 g ( 0.01 mol) , 室温下搅拌过夜, 乙酸乙酯 2 000 mL 稀释, 饱和碳酸氢钠溶液 ( 300 mL×2) , 饱和食盐水 (300 mL ×2) 洗涤, 干燥, 真空浓缩, 得到的粗品柱层析纯化 ( 乙酸乙酯-石油醚=3∶2 ) 得到 (6) 93.4 g,收率 75.7%。
[ 2R- (2α, 3β, 5α)] -5- [ 2- [ [ ( 4-甲氧基苯基) -二苯甲基] 氨基] -6- ( 苯甲氧基) -9H-嘌-9-基] -3- (苯甲氧基) -2- [ ( 苯甲氧基) 甲基] 环戊酮 ( 7) 的制备
在氮气保护下, 往干燥的三口反应瓶中加入 DMP 63.6 g (0.15 mol) 和二氯甲烷 ( 1 600mL) , 叔丁醇 12.2 mL ( 0.13 mol), 室温反应 10 min, 加入 (6) 82.3 g (0.1 mol) 的二氯甲烷 800 mL 溶液, 搅拌反应 30 min。 HPLC 监测反应, 反应完毕, 搅拌下加入混合溶液 (10%亚硫酸钠溶液-饱和碳酸氢钠溶液-饱和食盐水 =1.5∶1∶1) (3 500 mL), 搅拌 1 h。 分出有机相, 水相用二氯甲烷 1 600 mL 萃取, 合并有机相, 然后用饱和食盐水洗涤, 无水硫酸镁干燥, 浓缩至干得到黄色固体 ( 7) 80.0 g 。收率 97.4%。无需纯化, 直接用于下一步反应。
[ 1S- (1α, 3α, 4β)] -N- [ ( 4-甲氧基苯基) -二苯甲基] -6- ( 苯甲氧基) -9- [ 2-亚甲基-4-苯甲氧基-3- [ (苯甲氧基)甲基]环戊基] -9H-嘌呤-2-胺 (8) 的制备
将上述制备的 ( 7) 80.0 g( 0.097 mol) 的二氯甲烷 800 mL 溶液加入到烯化试剂( 1 600 mL) 中, 搅拌反应 3 h。 反应完毕, 把反应体系倒入二氯甲烷 (4 000 mL) 和饱和碳酸氢钠溶液 ( 4 000 mL) 混合体系中, 搅拌 1 h, 过滤, 滤饼用二氯甲烷洗涤。 分液,水相用二氯甲烷萃取 (1 500 mL), 合并有机相, 无水硫酸镁干燥。 浓缩至干, 得到粗产物柱层析纯化 (石油醚-乙酸乙酯=4∶1) , 得到泡沫状固体 ( 8) 39.5 g。 收率 49.5%。
[ 1S- ( 1α, 3α, 4β)] -2-氨基-1, 9-二氢-9- [ 2-亚甲基-4-苯甲氧基-3- [ (苯甲氧基) 甲基] 环戊基] -6H-嘌呤-6-酮( 9) 的制备
将 (8) 39.5 g (48.2 mmol) 溶于混合溶液 ( 四氢呋喃-甲醇 =1∶1) 800 mL 中, 加入 3 mol · L -1 盐酸 ( 197.5mL), 控温 60 ℃搅拌反应直到原料完全消失。 冷却至室温,加入水800 mL, 乙酸乙酯 800 mL, 搅拌下用 2 mol· L -1 氢氧化钠溶液调节 pH =7.1~ 7.3, 分液, 水相用乙酸乙酯( 800 mL ×2) 萃取, 合并有机相, 无水硫酸镁干燥, 浓缩至约 50 mL, 放置在冰箱中冷却过夜, 过滤收集固体, 固体用冷的乙酸乙酯洗涤。 40 ℃真空干燥, 得到白色固体 ( 9)19.8 g 。 收 率 89.8%。
2-氨基-1, 9-二氢-9- [ ( 1S, 3R, 4S) -4-羟基-3-羟甲基-2-亚甲基环戊基] -6H-嘌呤-6-酮一水合物 (1) 的制备
氮气保护下, 往干燥的三口瓶中加入 (9) 19.8 g ( 43.3mmol) , 干燥的二氯甲烷 ( 800 mL), 冷却至-70 ℃, 缓慢滴加三氯化硼 433.0 mL (433.0 mmol) , 滴加后继续搅拌反应 1 h, 缓慢升温至-30~ -20 ℃, 反应 1.5 h。 反应完毕,再降温至-70 ℃, 慢慢滴加甲醇 (400 mL) , 滴加完后升温至室温, 减压蒸除溶剂, 再加入甲醇 ( 800 mL) , 混合均匀后减压浓缩, 残渣用水 (1 200 mL) 溶解, 用乙醚萃取至乙醚层无色。 水相用 2 mol· L-1 氢氧化钠溶液调节 pH =7.0,然后将水相减压浓缩至 250 mL, 加活性炭 2.5 g 煮沸脱色30 min, 滤去活性碳, 滤液自然冷却, 室温放置过夜。 过滤, 冷蒸馏水洗涤, 蒸馏水重结晶, 得到白色晶体 (1) 8.2g 。 收率 64.2%。
参考文献
[1] Bisacchi GS, Chao ST , Bachard C, et al.BMS-200475, a novel carbocyclic-2′-deoxyguanosine analog with potent and selective anti-Hepatitis B Virus activity in vitro [J] .Bioo rg M ed ChemLett, 1997, 7 ( 2) :127-132.