背景及概述[3]
氮杂环丁烷类化合物作为医药中间体被大量使用得到重视。近年来通过各种官能团修饰已经成功的制备并试验了大量新化合物其中许多化合物具有良好的药物活性。如1-二苯甲基-3-氨基氮杂环丁烷和1-二苯甲基-3-羟基-3-氨甲基氮杂环丁烷近年来已应用在一些抗菌性、抗抑郁类药物合成中。1-叔丁氧羰基-3-胺基环丁胺也是其中重要的药物中间体应用于IRAK-4类抑制剂的合成。

制备[1-3]
报道一、
3-羟基-氮杂环丁烷-1-甲酸叔丁酯
将1.8g(7.52mmol)从制备例1-83-1得到的化合物溶于35ml甲醇中。向该溶液中加入1.81g(8.27mmol)二碳酸二叔丁酯、0.8g(7.9mmol)三乙胺、和吸附在活性炭上的钯(Pd/C)(10%,0.18g),并在氢气条件下搅拌16小时。通过硅藻土过滤反应混合物,在真空中蒸馏以除去溶剂,并使用己烷和乙酸乙酯的1∶1混合溶液,通过柱色谱法纯化,得到标题化合物1.06g(80%)。
1HNMR(400MHz,CDCl3);δ4.58(1H,m),4.15(2H,dd),380(2H,dd),2.08(1H,d),1.44(3H,s)
3-叠氮基-氮杂环丁烷-1-甲酸叔丁酯
将1.06g(6.12mmol)从制备例1-83-2得到的化合物和2.0g(7.65mmol)三苯基膦溶于40ml四氢呋喃中。向该溶液中加入1.61g(7.96mmol)偶氮二甲酸二异丙酯和2.1g(7.96mmol)二苯基磷酰基叠氮化物,并搅拌16小时。在真空中蒸馏反应混合物以除去溶剂,并使用己烷和乙酸乙酯的9∶1混合溶液,通过柱色谱法纯化,得到标题化合物1.21g(99%)。
1HNMR(400MHz,CDCl3);δ4.25~4.14(3H,m),3.89(2H,m),1.44(9H,s)
1-叔丁氧羰基-3-胺基环丁胺
将1.29g(6.51mmol)从制备例1-83-3得到的化合物溶于20ml甲醇中。向该溶液中加入吸附在活性炭上的钯(Pd/C)(10%,0.13g),并在氢气条件下搅拌3小时。通过硅藻土过滤反应混合物,在真空中蒸馏以除去溶剂,并使用甲醇和二氯甲烷的10∶90混合溶液,通过柱色谱法纯化,得到标题化合物0.82g(73%)。
1HNMR(400MHz,CDCl3);δ4.14(2H,dd),3.76(1H,m),3.58(2H,dd),1.44(9H,s)
报道二、
羟基-氮杂环丁烷-1-羧酸叔丁酯
将1-(二苯基甲基)-3-羟基氮杂环丁烷(300mg,1.26mmol,Oakwood,)溶解在乙酸乙酯(10mL)中。向其加入10%披钯碳(100mg)和二碳酸二叔丁酯(329mg,1.51mmol,Aldrich),将混合物在50psi下氢化过夜。过滤和去除溶剂提供期望产物。218mg,100%。MS(ES)MH+=174。
3-甲磺酰基氧基-氮杂环丁烷-1-羧酸叔丁酯
将醇(1.0g,5.77mmol,羟基-氮杂环丁烷-1-羧酸叔丁酯)溶解在二氯甲烷(20mL)中。向其中加入三乙胺(1.60mL,11.54mmol)和甲磺酰氯(536uL,5.77mmol,Aldrich),将混合物在0℃下搅拌3小时。去除溶剂,将残渣在乙酸乙酯和水之间分配。分离有机层并用硫酸钠干燥,浓缩以提供白色固体。1.51g,100%。将该化合物直接用于下一步骤。
3-叠氮基-氮杂环丁烷-1-羧酸叔丁酯
向搅拌的3-甲磺酰基氧基-氮杂环丁烷-1-羧酸叔丁酯(930mg,3.70mmol)在二甲基甲酰胺(20mL)中的溶液加入叠氮化钠(962mg,14.8mmol,Aldrich),将混合物在90℃下搅拌过夜。将混合物倒入水中,用乙酸乙酯(5×10mL)萃取,合并萃取液,用水、盐水洗涤,用硫酸钠干燥。去除溶剂以提供透明油。710mg,97%。将该化合物直接用于下一步骤。
1-叔丁氧羰基-3-胺基环丁胺
向搅拌的3-叠氮基-氮杂环丁烷-1-羧酸叔丁酯(1.15g,5.80mmol)在乙醇(20mL)中的溶液加入10%披钯碳(93mg),将混合物在48psi下氢化1小时。将混合物滤过硅藻土垫,去除溶剂至无色油。940mg,94%。
报道三、
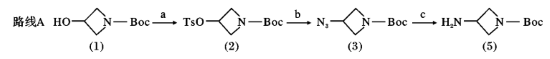
1-叔丁氧羰基-3-羟基氮杂环丁烷(500ml)、对甲苯磺酰氯(500ml)、氯化铵(500g)、水合肼(500ml)均为分析纯;邻苯二甲酰亚胺钾盐(500g)、叠氮化钠(100g)、锌粉(100g)均为化学纯;其他均为实验室常用试剂。所有试剂均按照试剂手册进行预处理。
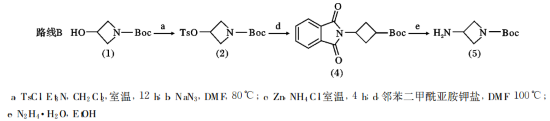
化合物(2)的合成
将化合物(1)2.08g(0.012mol)溶解于60ml二氯甲烷中冰浴下加入三乙胺6.65ml(0.048mol)然后分3次加入对甲苯磺酰氯9.12g(0.048mol)逐渐升温至室温反应反应12hTLC跟踪反应完成后将反应液冷却至室温加入40ml浓盐酸和200ml冰水的混合溶液用乙醚萃取3×50ml有机相用无水硫酸钠干燥柱层析得白色固体3.7g产率94.4%。
化合物(3)的合成
将化合物(2)2.5g(0.008mol)叠氮钠3g(0.048mol)溶解在20mlDMSO中升温至80℃反应搅拌4h。TLC跟踪反应完成后冷却至室温加入100ml乙醚有机相用水洗3×50ml饱和食盐水3×50ml无水无水硫酸钠旋蒸柱层析得白色液体1.48g产率93%。
化合物(3)~化合物(5)的合成
将化合物(3)1.39g(0.007mol)氯化铵1.5g(0.028mol)溶解在30ml含水的四氢呋喃溶液中搅拌下分批加入锌粉0.91g(0.014mol)室温下反应4h。TLC跟踪反应完成后加入50ml乙酸乙酯和5ml氨溶液过滤滤液用饱和食盐水洗涤无水硫酸钠干燥柱层析得无色液体0.7g产率70%。
化合物(4)的合成
将化合物(2)1g(0.003mol)邻苯二甲酰亚胺钾盐1.7g(0.009mol)溶解在15mlDMF中升温到100℃反应4h。TLC跟踪反应完成后减压蒸去DMF加入20ml二氯甲烷过滤滤液用饱和食盐水洗涤无水硫酸钠干燥柱层析得白色固体0.7g产率77%。
化合物(4)~化合物(5)的合成
将化合物(4)0.7g(0.002mol)2ml85%水合肼溶解在15ml无水乙醇中升温回流TLC跟踪反应完成后加入冰的稀盐酸淬灭反应并调节pH至5二氯甲烷洗涤水相3次用饱和碳酸氢钠水溶液调节pH至7二氯甲烷萃取3次合并有机相旋蒸得无色液体0.25g,产率63%。
参考文献
[1][中国发明]CN200980134833.4稠合杂环化合物
[2][中国发明]CN200480003929.44-氨基嘧啶-5-酮
[3]吴孝增,崔杨.氮杂环丁烷类药物中间体的合成[J].应用化工,2010,39(4):618-619,622.DOI:10.3969/j.issn.1671-3206.2010.04.043.