背景及概述[1][2]
自1962年萘啶酸问世以来,人们已经合成了数以万计的喹诺酮类抗菌药并对其活性进行了评估。在母核(喹诺酮核或1,8-萘啶酮核)的基础上,通过结构改造如1-位引入取代基(乙基、环丙基、氟代苯基等),6-位引入氟原子、7-位引入环胺基(哌嗪基、3-氨基吡咯烷基等)、8-位引入甲氧基等,使这类药物的抗菌谱、抗菌活性和药代动力学性质等不断得到改善。
目前,喹诺酮类抗菌药已发展至第四代,主要代表药物有:司帕沙星、格帕沙星、妥舒沙星、加替沙星、曲伐沙星、莫西沙星、克林沙星、吉米沙星、佳诺沙星等。与第三代喹诺酮类抗菌药相比,第四代喹诺酮类抗菌药在保持了原有对革兰氏阴性菌的良好抗菌活性的基础上,明显增强了对革兰氏阳性菌(如金黄色葡萄球菌、肺炎链球菌等)的抗菌活性,已成为目前临床上治疗各系统感染尤其是呼吸道感染的有力武器。
克林沙星(Clinafloxacin,CF),化学名为7‑(3‑氨基‑1‑吡咯烷基)‑1‑环丙基‑6‑氟‑8‑氯‑1,4‑二氢‑4‑氧代‑3‑喹啉羧酸(结构式如式A所示),是美国沃纳‑兰博(Warner‑Lambert)药物公司开发的抗菌药,在国外已完成三期临床评价,曾经由于具有优秀的广谱抗菌活性尤其是对革兰阳性菌、厌氧菌和肺炎支原体具有高活性而倍受关注,但因临床使用发现存在严重的毒性问题而停止研发。此外,克林沙星在制药方面还存在溶解性差等问题。
Warner-Lambert公司已中止了喹诺酮类抗生素克林沙星(clinafloxacin)的开发,并放弃了其用于医院内严重感染治疗的美国上市申请。在过去的一年里,FDA对本品可能的不良反应,如肝脏毒性和心律失常等表示关注,而这些不良反应难以回顾性地从临床研究资料中得到。静注给药的本品还有光毒性和低血糖等不良反应,由此限制了其在院内的使用,而且增加的这些不良反应还可影响本品的风险-效益评估。
应用[1][3]
克林沙星(clinafloxacin)是目前正在研制的第四代喹诺酮类抗菌药,是一种新的广谱抗菌药,它抑制细菌的DNA促旋酶,其作用和疗效比目前临床上广泛使用的喹诺酮类抗菌药好,它的动物毒性与喹诺酮类相似,因而在临床上具有较高的使用价值。其化学结构为在母环的8位上引入了氯原子,7位哌嗪环上有氨基,其结构式如下:
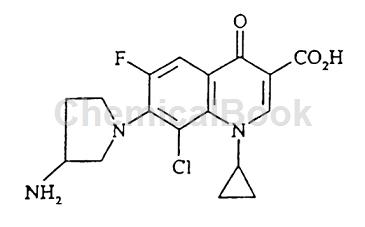
克林沙星
克林沙星是目前抗菌谱广、抗菌活性非常强的“超级抗菌药物”,对禽类的大肠杆菌感染、支原体引起的呼吸道感染、巴氏杆菌引起的鸭浆膜炎有特效。在动物的疾病防治上,尤其是食品动物,克林沙星具有广阔的应用前景。
克林沙星(CF)曾经是第四代喹诺酮类抗菌新品种中具有“超广谱”性质的明星分子。大量体外和体内研究发现,CF不仅具有超强的抑制杀灭作用,同时显示生物利用度高、组织穿透力强、安全性和耐受性好、血清半衰期长及药代动力学性质优良等药学性质。研发单位曾将克林沙星推向三期临床,然而克林沙星固有的光毒性、心脏毒性及低血糖发生率等不良反应,妨碍了其上市申请。此外,实验过程中发现CF原药溶解性较小,水溶液中的稳定性不够,可能也是影响其成药的重要原因。
克林沙星的衍生物,最简单可行的策略就是保留克林沙星的母体结构、将其非药效基团进行结构修饰。而众多研究表明,喹诺酮的7位取代基能够很大程度上影响DNA拓扑异构酶合成及细胞通透性,并最终影响其生物活性、抗菌谱、溶解度和药代动力学。发明人对克林沙星7位吡咯环上3-氨基进行修饰衍生,合成了200多个克林沙星氨基衍生物,包括在氨基上引入五元或六元杂环、脲和胺等,从中获得了毒性降低、活性增强、溶解性提高和分子稳定性增加的抗菌活性分子。
对克林沙星进行了杀菌试验研究,发现克林沙星为一较强的杀菌剂,其MBC值约为MIC值的4~16倍,对金黄色葡萄球菌和大肠埃希菌的杀菌时间约在8h左右。克林沙星对有害菌总的MIC50为0.03~1.0mg·L-1,远远低于单次口服200mg克林沙星在血浆中的峰浓度(2.5mg·L-1),其MIC90值除嗜麦芽窄食单胞菌外(4mg·L-1),亦皆在2.5mg·L-1以下,说明克林沙星对临床上常见致病菌在常规剂量下,均有较好的抗菌活性。克林沙星原料药对常见的致病菌金黄色葡萄球菌,大肠埃希菌,铜绿假单胞菌,肠球菌,肺炎克雷伯菌和福氏志贺氏菌引起的小鼠感染有确切的治疗作用,其ED50较对照药环丙沙星为低,说明克林沙星的体内抗菌作用亦明显优于环丙沙星。
克林沙星衍生物制备[2]
1. 克林沙星氨基衍生物
向反应瓶中加入克林沙星(CF)、K2CO3及适量二氯甲烷(DCM),在冰浴下滴加氯乙酰氯 的DCM溶液,CF、K2CO3和氯乙酰氯的投料摩尔比为10:25:15~25,滴毕,冰浴下搅拌反应, TLC(薄层色谱法)监测反应进程。反应完毕,得到黄绿色浑浊液,抽滤,滤饼用DCM洗涤, 洗液与滤液合并,旋蒸至干,得中间体IM1。
向反应瓶中加入胺组分、K2CO3及催化量的KI和适量溶剂,室温搅拌30分钟后加入中 间体IM1,IM1、K2CO3与胺组分的投料摩尔比为1:3:2,30-45℃控温反应,TLC监测反应进 程。
反应完毕,根据具体情况分别按以下几种方式进行后处理:(1)若产物溶于有机溶剂, 加水,用2N HCl调pH至6-7,用DCM萃取,收集有机相,旋蒸得粗品,用二氯甲烷-石油 醚(DCM-PE)重结晶;(2)若产物溶于水,调pH至中性,静置分层,收集水相,继续调pH至 4~5,冷藏析晶,抽滤,滤饼用90%乙醇重结晶;(3)若反应体系有固体产生,抽滤,滤饼加 水,用2N HCl调pH至4~5,冷藏析晶;(4)若产物既溶于有机溶剂,又溶于水,先旋蒸除 去有机相,再用2N HCl调pH至4~5,冷藏析晶,即得1.克林沙星氨基衍生物。
2. 胺基烷酰克林沙星衍生物
在100mL圆底烧瓶中依次加入原料CF、K2CO3及适量二氯甲烷(DCM),在冰浴下滴加 氯乙酰氯的DCM溶液,CF、K2CO3与氯乙酰氯的投料摩尔比为1:2.5:1.5~2.5,滴毕,冰浴下 持续搅拌反应,TLC(薄层色谱法)监测反应进程。反应完成后,得到黄绿色浑浊液,抽滤,滤 饼用DCM洗涤,洗液与滤液合并,旋蒸得粗品,其纯度满足后续反应需要。将粗品经柱层 析纯化得纯品,干燥,得到中间产物IM1
向反应瓶中加入原料胺组分、K2CO3及催化量的KI和适量溶剂,室温搅拌30分钟后, 加入IM1;IM1、K2CO3与胺组分的投料摩尔比为1:3:2,室温(10℃-40℃)搅拌反应,TLC监 测反应进程。
反应结束后,根据具体情况分别按以下几种方式进行后处理:(1)若产物溶于 有机溶剂,加水,用2N HCl调pH至6-7,用DCM萃取,收集有机相,旋蒸得粗品,用二 氯甲烷-石油醚(DCM-PE)重结晶;(2)若产物溶于水,调pH至中性,静置分层,收集水相, 继续调pH至4~5,冷藏析晶,抽滤,滤饼用90%乙醇重结晶;(3)若反应体系有固体产生, 抽滤,滤饼加水,用2N HCl调pH至4~5,冷藏析晶;(4)若产物既溶于有机溶剂,又溶于 水,先旋蒸除去有机相,再用2N HCl调pH至4~5,冷藏析晶,即得胺基烷酰克林沙星衍生物。
3. 氯甲酰化克林沙星衍生物
向500mL反应瓶中依次加入CF 18.5g(50mmol)和氯仿150mL,搅拌均匀后,缓慢滴 加BTC 5.2g(17.5mmol)的氯仿溶液。滴加完毕后,冰浴冷却,剧烈搅拌,持续反应7小时后, 缓慢滴加TEA 30mmol,滴加完毕,室温(22℃)下反应,薄层色谱法(TLC)监测反应进程。反 应完毕(约0.5h),抽滤,滤饼用二氯甲烷(DCM)洗涤后,滤液加水100mL,用2N HCl调pH 至4~5,再用DCM(2×100mL)萃取2次,收集有机相,用饱和食盐水(2×100mL)洗涤,无水 硫酸钠干燥,过滤,滤液旋蒸浓缩,快速柱层析法纯化,得中间体IM120.220g,收率94.7%。
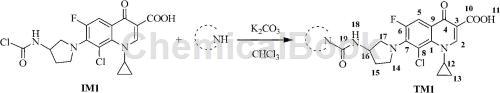
向100mL反应瓶中依次加入氯仿4mL、胺1.5mmol、碳酸钾2mmol,室温搅拌0.5小 时后,加入原料IM11mmol,控温反应,TLC监测反应进程;反应完毕后,再根据具体情况 分别按以下几种方式进行后处理:(1)对于水溶性较差的产物,抽滤,滤饼用DCM洗涤后, 加水,调pH至弱酸性,直接用DCM萃取,收集有机相,用DCM‑PE(石油醚)重结晶;(2) 对于水溶性较好的产物,调pH至弱酸性,加入固体食盐至达到饱和,再用DCM萃取,收集 有机相,用DCM‑PE(石油醚)重结晶;或者,调pH至弱酸性,加入固体食盐至达到饱和,置 冰箱中冷藏析晶,次日抽滤,即得纯品。
主要参考资料
[1]童明庆, & 戴传箴. (2000). 克林沙星在体内外的抗菌活性. 中国医院药学杂志, 20(8), 463-465.
[2]黄山, & 李泽. (2000). 克林沙星的2,4,5-三氟苯甲酸路线合成. 中国医药工业杂志, 31(8), 338-340.
[3]周梅华, 郁韵秋, 段更利, 程务本, 许长江, & 刘骁. (2001). 克林沙星在大鼠体内的药代动力学和生物利用度. 药学学报, 36(2), 134-136.