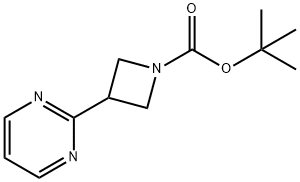
tert-butyl 3-(pyriMidin-2-yl)azetidine-1-carboxylate synthesis
- Product Name:tert-butyl 3-(pyriMidin-2-yl)azetidine-1-carboxylate
- CAS Number:1236861-59-8
- Molecular formula:C12H17N3O2
- Molecular Weight:235.28
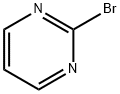
4595-60-2
354 suppliers
$8.00/5g
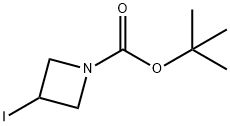
254454-54-1
261 suppliers
$15.00/1g
1236861-59-8
7 suppliers
inquiry
Yield:1236861-59-8 31%
Reaction Conditions:
Stage #1: tert-butyl-3-iodoazetidine-1-carboxylatewith chloro-trimethyl-silane;ethylene dibromide;zinc in tetrahydrofuran at 20 - 50;Molecular sieve;
Stage #2: 2-bromopyrimidine;tetrakis(triphenylphosphine) palladium(0) in tetrahydrofuran; for 25 h;Molecular sieve;
Steps:
6.2.A
Zinc powder (150.1 g, 2.30 mol) and molecular sieves (50 g) were combined in a reaction flask and flame-dried under vacuum for 10 mins. Once the flask had returned to room temperature, it was charged with THF (4 L), and 1,2-dibromoethane (24.4 mL, 0.28 mol) was added. The reaction mixture was heated to 50° C. for 10 mins, then allowed to come to ambient temperature, at which time trimethylsilyl chloride (33.5 mL, 0.264 mol) was added {Caution: slightly exothermic}. The mixture was allowed to stir at room temperature for about 18 h. Slow addition of C7 (500 g, 1.77 mol) over 1.5 h was followed by stirring for an additional 18 h. In a separate flask, 2-bromopyrimidine (253 g, 1.59 mol) was combined with molecular sieves (85 g) in THF (1.3 L), and the mixture was degassed. This mixture was treated with tetrakis(triphenylphosphine)palladium(0) (32.7 g, 0.0283 mol), then added to the flask containing the reaction mixture from C7. The reaction was stirred for 25 h, and then filtered through Celite. The filtrate was concentrated under reduced pressure, then partitioned between saturated aqueous sodium carbonate solution (2 L) and EtOAc (2 L). The aqueous layer was extracted with EtOAc (2×2 L), and the combined organic layers were dried over sodium sulfate and concentrated in vacuo. The resulting yellow liquid residue was triturated with methyl tert-butyl ether (500 mL), and the precipitate was removed by filtration. Partial concentration of the filtrate resulted in precipitation of a solid; the mixture at this point was cooled in an ice-water bath. Filtration then provided a solid, which was washed with a minimum quantity of cold methyl tert-butyl ether to afford C33 as a white solid, which was taken directly into the next step. Yield: 131 g, 0.557 mol, 31%. GCMS m/z 180 ([M-tert-butyl]+1); 136 ([M-BOC]+1). 1H NMR (300 MHz, CDCl3) δ 1.45 (s, 9H), 4.0 (m, 1H), 4.3 (m, 4H), 7.2 (t, 1H), 8.75 (d, 2H).
References:
US2010/190771,2010,A1 Location in patent:Page/Page column 20-21
141699-58-3
112 suppliers
$6.00/250mg
1236861-59-8
7 suppliers
inquiry
141699-55-0
390 suppliers
$6.00/1g
1236861-59-8
7 suppliers
inquiry