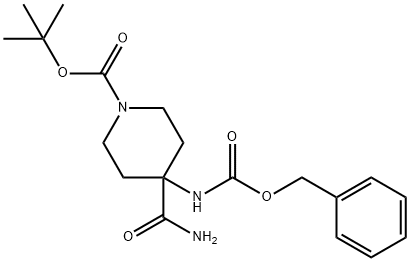
tert-butyl 4-(benzyloxycarbonylamino)-4-carbamoylpiperidine-1-carboxylate synthesis
- Product Name:tert-butyl 4-(benzyloxycarbonylamino)-4-carbamoylpiperidine-1-carboxylate
- CAS Number:288154-17-6
- Molecular formula:C19H27N3O5
- Molecular Weight:377.43
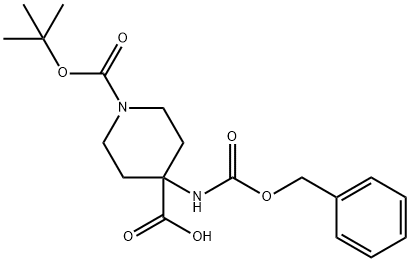
288154-16-5
288154-17-6
General procedure for the synthesis of tert-butyl 4-(benzyloxycarbonylamino)piperidine-1,4-dicarboxylate from mono-tert-butyl 4-benzyloxycarbonylaminopiperidine-1,4-dicarboxylate: firstly, 4-(benzyloxycarbonylamino)-1-(tert-butoxycarbonyl)piperidine-4-carboxylic acid (1A, 2.0 g, 1.0 eq.) was solubilized in DME (0.5 M), NMM (1.0 eq.) and IBCF (1.0 eq.) and reacted at -15 °C for 10 min. Subsequently, ammonia (1.5 eq.) was added and the reaction mixture was stirred at room temperature for 1.5 hours. After completion of the reaction (confirmed by LCMS analysis), it was partitioned with ethyl acetate and water. The organic phase was washed with brine, dried over Na2SO4, filtered and the solvent was removed under reduced pressure. The residue was purified by ether crystallization to afford tert-butyl 4-(benzyloxycarbonylamino)-4-carbamoylpiperidine-1-carboxylate (1B, 70% yield). 1B (1.0 equiv) and a catalytic amount of acetic acid were dissolved in MeOH (0.05 M) and hydrogenated at 50 °C through an H-cube hydrogenator equipped with a Pd/C column. After completion of the reaction, methanol was removed under reduced pressure and the product was washed twice with cold ether to afford tert-butyl 4-amino-4-carbamoyl piperidine-1-carboxylate (1C, 90% yield). 1C (0.3 M) was mixed with 2-chloro-1,1,1-trimethoxyethane (4.0 eq.) and acetic acid (2.0 eq.) and stirred at 118 °C for 12 hours. After removing the solvent under reduced pressure, the residue was purified by crystallization from cold ether to afford tert-butyl 2-(chloromethyl)-4-oxo-1,3,8-triazaspiro[4.5]dec-1-ene-8-carboxylate (1D, 67% yield). 1D was dissolved in 2.0 M ammonia in 2-propanol solution (0.04 M), an excess of ammonia was drummed into a sealed tube and heated at 60 °C for 12 hours. Upon completion of the reaction (confirmed by LCMS analysis), the solvent was removed to afford tert-butyl 2-(aminomethyl)-4-oxo-1,3,8-triazaspiro[4.5]dec-1-ene-8-carboxylate (1E), which did not require any further purification. eSI-MS: m/z 283.1 (M+H)+. For the deprotection step, 1E (1.0 eq.) was dissolved in CH2Cl2 and a 1.6:1 solution of CH2Cl2/TFA (final concentration 0.15 M) was slowly added at 0 °C. The reaction was completed within 30 min at 0°C (confirmed by LCMS analysis). Volatiles were removed under reduced pressure to afford the TFA salt of 2-(aminomethyl)-1,3,8-triazaspiro[4.5]dec-1-en-4-one (1G), which was ready for use without further purification.ESI-MS: m/z 183.1 (M+H)+.
288154-16-5
99 suppliers
$34.00/250mg
288154-17-6
21 suppliers
inquiry
Yield:288154-17-6 70%
Reaction Conditions:
Stage #1: 4-(Benzyloxycarbonylamino)-1-(tert-butoxycarbonyl)-piperidine-4-carboxylic Acidwith 4-methyl-morpholine;isobutyl chloroformate in 1,2-dimethoxyethane at -15;
Stage #2: with ammonia in 1,2-dimethoxyethane at 20;
Steps:
1
A solution 4-(benzyloxycarbonylamino)-1-(tert-butoxycarbonyl)piperidine-4-carboxylic acid (1A, 2.0 g, 1.0 equivalent) in DME (0.5 M) was treated with NMM (1.0 equivalent) and IBCF (1.0 equivalent) at -15° C. After 10 min, aqueous ammonia (1.5 equivalents) was added. The reaction mixture was stirred at room temperature for 1.5 hr. The reaction was complete as determined by LCMS analysis and then partitioned between ethyl acetate and water. The organics were subsequently washed with brine, then dried over Na2SO4, filter, and the volatiles removed under reduced pressure. The residue was purified by crystallization from diethyl ether to afford the tert-butyl 4-(benzyloxycarbonylamino)-4-carbamoylpiperidine-1-carboxylate (1B, 70%).1B (1.0 equivalent) and a catalytic amount of acetic acid in MeOH (0.05M) was passed through H-cube hydrogenator equipped with Pd/C cartridge at 50° C. The methanol in the reaction mixture was then removed under reduced pressure, and the product was washed with cold ether twice to afford the tert-butyl 4-amino-4-carbamoylpiperidine-1-carboxylate (1C, 90%).1C (0.3M) was added 2-chloro-1,1,1-trimethoxyethane (4.0 equivalents) and acetic acid (2.0 equivalents). The mixture was stirred for 12 hr at 118° C. Solvent was removed under reduced pressure and the residue was purified by crystallization from cold diethyl ether to afford tert-butyl 2-(chloromethyl)-4-oxo-1,3,8-triazaspiro[4.5]dec-1-ene-8-carboxylate (1D, 67%).1D in a seal tube was added 2.0 M ammonia in 2-propanol (0.04M). Excess ammonia gas was bubbled in and the mixture was heated at 60° C. for 12 hr. The reaction was complete as determined by LCMS analysis. After removal of solvent; the residue tert-butyl 2-(aminomethyl)-4-oxo-1,3,8-triazaspiro[4.5]dec-1-ene-8-carboxylate (1E) was used without further purification. ESI-MS: m/z 283.1 (M+H)+.For deprotection, 1E (1.0 equivalent) was dissolved in CH2Cl2 at 0° C., and slowly treated with a 1.6:1 solution of CH2Cl2/TFA (2:1 final ratio, 0.15 M). The reaction was completed in 30 minutes at 0° C. as determined by LCMS analysis. The volatiles were removed under reduced pressure to yield the respective 2-(aminomethyl)-1,3,8-triazaspiro[4.5]dec-1-en-4-one (1G) as TFA salts which were used without further purification. ESI-MS: m/z 183.1 (M+H)+.
References:
US2009/253725,2009,A1 Location in patent:Page/Page column 51