133454-47-4
中文名称
伊潘立酮
英文名称
ILOPERIDONE
CAS
133454-47-4
分子式
C24H27FN2O4
MDL 编号
MFCD00866688
分子量
426.48
MOL 文件
133454-47-4.mol
更新日期
2026/07/16 12:23:04
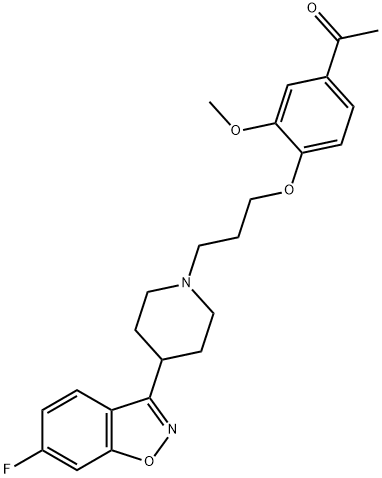 133454-47-4 结构式
133454-47-4 结构式
基本信息
中文别名
伊潘利酮伊潘立酮
20MG/100MG/1KG
1-[4-[3-[4-(6-氟-1,2-苯并异噁唑-3-基)-1-哌啶基]丙氧基]-3-甲氧基苯基]乙酮
英文别名
HP 873Fanapt
ZoMaril
ILOPERIDONE
Iloperidone-P88
Iloperidone(Fanapt)
Iloperidone solution
Iloperidone(ILO-522,HP-873)
1,2-Benzisoxazole,ethanone deriv.
Iloperidone P88 metabolite-13DD3 (Racemic)
所属类别
原料药:抗精神病药物理化学性质
熔点118-120°C
沸点593.7±50.0 °C(Predicted)
密度1.204±0.06 g/cm3(Predicted)
闪点9℃
储存条件-20°C Freezer, Under Inert Atmosphere
溶解度DMSO:可溶,5mg/mL,澄清
酸度系数(pKa)8.43±0.20(Predicted)
形态粉末
颜色白色至米色
Merck14,4900
InChI1S/C24H27FN2O4/c1-16(28)18-4-7-21(23(14-18)29-2)30-13-3-10-27-11-8-17(9-12-27)24-20-6-5-19(25)15-22(20)31-26-24/h4-7,14-15,17H,3,8-13H2,1-2H3
InChIKeyXMXHEBAFVSFQEX-UHFFFAOYSA-N
SMILESFc1cc2[o]nc(c2cc1)C3CCN(CC3)CCCOc4c(cc(cc4)C(=O)C)OC
安全数据

警示词危险
危险性描述H301-H413
危险品标志F,T
安全说明16-36/37-45
危险品运输编号UN2811 - class 6.1 - PG 3 - EHS - Toxic solids, organic, n.o.s., HI: all
WGK Germany1
RTECS号KM5777850
海关编码29349990
存储类别6.1C - Combustible acute toxic Cat.3
toxic compounds or compounds which causing chronic effects
toxic compounds or compounds which causing chronic effects
危险性类别Acute Tox. 3 Oral
Aquatic Chronic 4
Aquatic Chronic 4
常见问题列表
非典型抗精神病药物
伊潘立酮(iloperidone)是一种非典型抗精神病药物,为5-羟色胺-2(5-HT2)和多巴胺2(D2)双重拮抗剂,伊潘立酮的研发过程有点戏剧性,最初由赫斯特-罗素制药公司合成并鉴定,后来该公司决定放弃对其的进一步开发, 并将其转让给Titan制药公司;1997年1月, Titan制药公司又将本品二次转让给了诺华公司,后者投资本品的III期临床开发并承担除了日本(Titan制药公司仍保留本品在日本的开发权)以外的全球范围的本品注册费用;然而, Vanda制药公司于2004年又从诺华公司获得本品的开发权。2009年5月6日,美国FDA批准由美国Vanda Pharma公司开发成功伊潘立酮(商品名:Fanapt)上市,临床上主要用于治疗成年人精神分裂症。伊潘立酮作为口服非典型抗精神病药物, 对减轻精神分裂患者的阳性症状(如幻觉、妄想、思维紊乱、敌视、怀疑) 和阴性症状( 如反应迟钝、情绪淡漠、社交淡漠、少语) 均有明显效果。伊潘立酮属于5-HT2 /D2 受体拮抗剂, 对多巴胺D3 受体也有很高的亲和力, 对肾上腺素a1受体、多巴胺D4 受体、5-HT6 和5-HT7 也具有适当的亲和力, 对5-HT1A、多巴胺D1和组胺H1受体有较低的亲和力。与目前使用的抗精神病药物比较, 短期、长期的安全试验结果显示, 伊潘立酮的不良反应要少些, 患者体质量增加幅度较小, 伊潘立酮不会诱导患者发生糖尿病, 患者锥体外系症状也较少( 如无静坐不能、无高催乳素血症、嗜睡发生率下降、认知能力下降较少等) 。临床研究结果显示, 伊潘立酮具有良好的安全性和耐受性, 代谢情况良好, 主要通过肝脏代谢排出体外。伊潘立酮最常见的不良反应为眩晕、口干、疲乏、鼻充血、体位性低血压、嗜睡、心率加快、体质量增加和QT间期延长等。伊潘立酮能增加老年痴呆性精神病患者的死亡危险, 所以老年痴呆性精神病患者不可使用此药。
以上信息由chemicalbook的晓楠编辑整理。
理化性质
本品为白色至灰白色结晶粉末, 在水中几乎不溶, 在盐酸中微溶, 在氯仿、乙醇、甲醇及乙腈中易溶。药理作用
伊潘立酮是一种哌啶基苯唑衍生物, 在啮齿类动物中, 对5-HT2受体具有较高的亲和力(IC50=9.3nmol/L) , 而对D2受体的亲和力则相对降低了一个数量级(IC50=109nmol/L); 对肾上腺素a1受体亦具极高的亲和力(IC50=0.4 nmol/L) , 但对a2、5-HT1A、O和D1受体的亲和力则低得多(IC50分别为60、210、180 和750nmol/L )。本品不与毒蕈碱乙酰胆碱受体和N-甲基-D-天门冬氨酸离子通道位点结合。体内外实验表明, 本品具有与其他非典型抗精神病药物一样的药理学特性, 即具对中脑边缘的选择性作用和抗精神病症状作用, 因而致低EPS发生率。
活体外受体放射自显影术研究发现, 连续19天给大鼠腹腔注射5mg/kg剂量的本品, 能够显著减少额叶皮质中5-HT2受体的数量, 但不影响受体亲和性, 而且也不影响伏隔核和纹状体6个区域中D2 受体的数量和亲和性。这是本品与氟哌啶醇明显不同之处, 后者能够增加这些区域D2受体的数量(这或许是其EPS发生率高的因素之一)。
在表达有人D2A受体和人a2C肾上腺素能受体的HEK-293和CHO-K1 细胞中考察本品对麦角二乙胺(LSD)激动剂活性影响的研究显示, LSD对D2A受体和a2C肾上腺素能受体的激动剂活性(pIC50分别为8.69 +/-0.08 和8.73+/-0.05) 均能被本品完全阻断[pKB分别为(8.684+/-0.14)和(8.13+/-0.03)]。所以, 对a2C肾上腺素能受体的阻断可能增强D2受体阻断所致的抗精神病作用。
放射配体结合试验显示, 本品对a1肾上腺素能受体以及D3和5-HT2A受体具高亲和力, 表明其为一潜在的具有较少副作用的广谱抗精神病药物。
合成方法
(1)以间二氟苯为起始原料, 经傅-克酰基化反应, 再与羟胺反应成肟, 在强碱氢化钠作用下发生分子内取代反应成环, 再与1-[4-(3-氯丙氧基)-3-甲氧基苯基]乙酮反应, 得到伊潘立酮。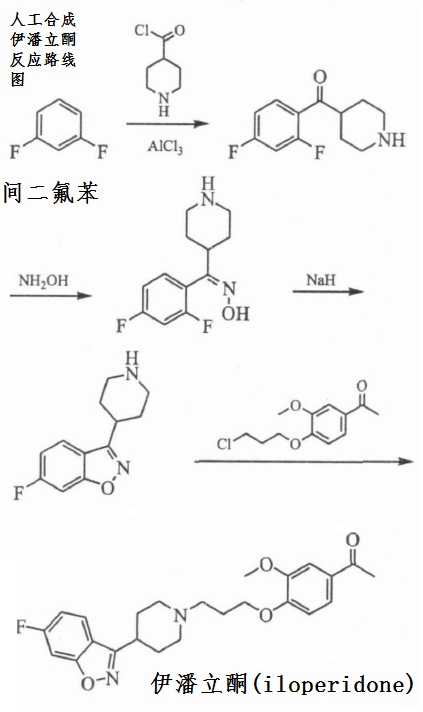
图1为人工合成伊潘立酮反应路线图。
(2)以间氟苯酚为起始原料, 经傅-克酰基化, 再与羟胺反应成肟,肟与乙酸酐发生取代反应后, 在氢化钠作用下发生分子内取代反应成环, 再与1-[4-( 3-氯丙氧基)-3-甲氧基苯基]乙酮反应, 得到伊潘立酮。
药物相互作用
1. 本品不是CYP1A1, CYP1A2, CYP2A6, CYP2B6,CYP2C8, CYP2C9, CYP2C19或CYP2E1的底物, 因此本品与这些酶的诱导剂或抑制剂合用时不会发生相互作用。本品主要通过CYP3A4 和CYP2D6 代谢。CYP3A4抑制剂(如酮康唑)或CYP2D6抑制剂(如氟西汀、帕罗西汀)抑制本品代谢并使血药浓度水平增加。2. 人体肝微粒体体内试验表明, 本品对CYP1A1,CYP1A2, CYP2A 6, CYP2B6, CYP2C8, CYP2C9或CYP2E1 酶代谢的药物无抑制作用, 对CYP1A2,CYP2C8, CYP2C9, CYP2C19, CYP3A4和CYP3A5酶未产生酶诱导效应。
3. 本品应避免与延长QT间期的药物合用。包括IA类抗心律失常药(奎尼丁、普鲁卡因胺)或III类抗心律失常药(胺碘酮、索他洛尔);抗精神病药物(氯丙嗪、硫利达嗪);抗菌药(加替沙星、莫西沙星);其他(戊双脒、左旋乙酰美沙酮)。本品应避免使用于先天性QT间期延长和有心律失常病史的患者。
不良反应
与目前使用的抗精神病药物比较, 短期、长期的安全试验结果显示, 本品的不良反应少, 患者体质量增加幅度较小, 不会诱导患者发生糖尿病, 患者锥体外系症状(如无静坐不能、无高催乳素血症、嗜睡发生率下降、认知能力下降较少等) 也较少。临床研究结果显示, 本品具有良好的安全性和耐受性, 代谢情况良好, 主要通过肝脏代谢排出体外。最常见的不良反应为眩晕、口干、疲乏、鼻充血、体位性低血压、嗜睡、心率加快、体重增加和QT 间期延长等。本品能增加老年痴呆性精神病患者的死亡危险, 所以老年痴呆性精神病患者不可使用此药。知识产权与专利
国内有关伊潘立酮的专利有两个,申请人均为瑞士诺瓦提斯公司,其一是伊潘立酮和星状聚合物的贮库制剂,2002年申请,至今未授权,属于特殊制剂剂型的组合物专利,与申报伊潘立酮及片无关;另一个是伊潘立酮的新用途专利,2002年申请,2005年授权,保护范围是伊潘立酮在包括双极型情感障碍在内的情感障碍的治疗中的用途,并不是精神分裂的用途,也不影响伊潘立酮及片的申报。