背景及概述[1]
甲烷-D4为甲烷的同位素衍生物,简称CD4。甲烷和甲烷-D4作为最简单的有机物之一,它们的分子光谱很早就有人作了实验研究,而且甲烷也一直是量子化学计算的重要算例。
分子几何构型[1]
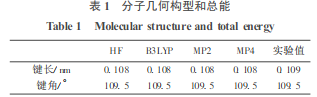
用 HF/6-31G **,B3LYP/6-31G**,MP2/6-31G**,MP4/6-31G**等方法对甲烷和甲烷-D4进行几何构型全优化,所得的 C-H, C-D 键长均为 0.1084 nm,H-C-H键角均为 109.5°,与实验结果吻合得很好(表1) 。
振动基频[1]
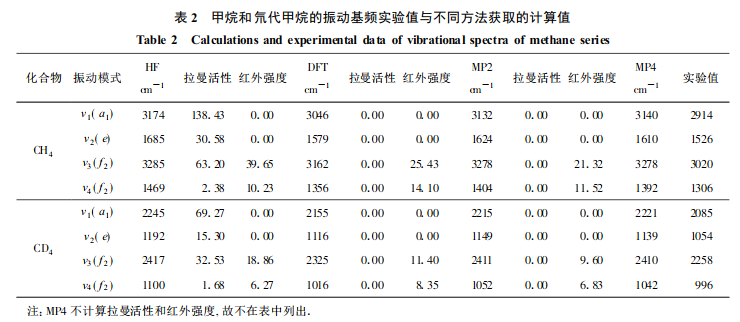
在用Gaussian 98 程序对甲烷和甲烷-D4的研究中,分别用电子自旋限制 Hatree-Fock方法、B3LYP密度泛函方法、二级微扰、四级微扰进行了计算,都采用6-31G**标准基组,计算所得的基频值如表 2 所示。从表 2 可见,计算结果与实验值差值由大到小依次为:HF >MP2 >MP4 >DFT/B3LYP。CH4 振动频率的计算值与实验值相差为12.4%( HF 计算的差),最小为 3.5%(B3LYP 计算的最小差)。CD4振动频率的计算值与实验值相差 10.4 %(HF 计算的差),最小为 2.0 %( B3LYP 计算的最小差)。同时,可以明显看出,由于氢原子被氘原子取代后分子振动频率改变。二级微扰、四级微扰方法考虑了电子相关能,因而结果有所改善,但同时也大幅度增加了机时。密度泛函理论将相关能表示为电子密度的泛函,简化了计算,而且相关作用对电子的密度影响小,故其结果较好且占用机时小于微扰方法。
热力学性质[1]
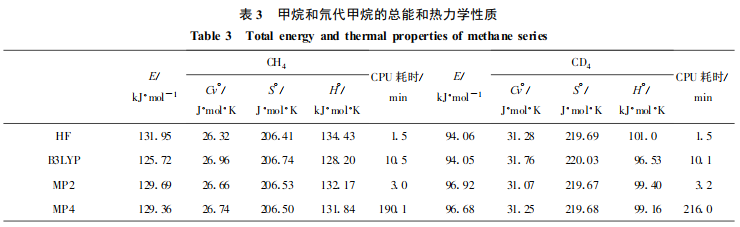
对进行几何构型全优化后得到的甲烷和甲烷-D4分子,用HF/6-31G**, B3LYP/6-31G**,MP2/6-31G**,MP4/6-31G**等方法计算总能和热力学性质,结果见表 3。氘取代氢原子后, 分子等容热容和熵增加而总能和焓却减小。
主要参考资料
[1]陆春海,陈文凯,廖俊生,王怀胜,汪小琳,孙颖.氘代甲烷几何构型及物性的量子化学研究[J].计算物理,2003(01):88-90.