概述【1】【2】
喹那普利是一种无巯基、长效口服的新型血管紧张素转换酶抑制剂。1989年应用于临床,1991年美国FDA批准上市用于治疗高血压和充血性心力衰竭。国产盐酸喹那普利片于1999年由上海医药工业研究院和哈药集团制药总厂联合研制成功并应用于临床,商品名为“益恒”。喳那普利属于第三代ACIE,它是由认值rner一Lambert公司开发的无琉基、长效、口服ACE抑制剂,1989年7月首次在英国和意大利上市,。1991年经美国FDA批准在美国上市治疗高血压和充血性心力衰竭。哇那普利是一个前体药物,其吸收后水解成更具有活性的二元酸ACE抑制剂一喳那普利拉。
与其它ACIE前体药物相比,抑制ACE活性与雷米普利拉相似,比培噪普利、依那普利、卡托普利强,由于喳那普利及其二元酸都具有抑制ACE活性,可降低血管紧张素和醛固酮的血浆浓度水平且增加强度激肤活性,哇那普利对肾血管型高血压降压效果比卡托普利、马来酸依那普利强,不良反应发生和因付作用停药的比例均低于卡托普利和马来酸依那普利,对肾毒性更轻。
哇那普利治疗高血压同时具有降血脂作用,治疗高血压有效剂量10mg一40mg/d,喳那普利平均消除半衰期为1小时,哇那普利拉半衰期为2小时,健康自愿者给药2.5一20mg在24小时内,血浆ACE活性被抑制70一95%,对AcE抑制作用可持续达48小时,ManazatoE报导了6003例中重度高血压患者(3004例男,2999例女),服用3一6个月盐酸哇那普利,舒张压从loZmmHg下降至87mmHg。盐酸哇那普利是第3代ACE抑制剂优秀代表之一,现名列全球畅销药的第91位,2000年销售额达5.53亿美元,它的研制成功提供了一个毒副作用小,因副作用需停药率低,对肾血管型高血压疗效好,兼有降血脂作用的新药,适合长期服用治疗高血压和心血管疾病。
药理毒理【3】
1.本品为无巯基、长效、口服血管紧张素转换酶(ACE)抑制剂,口服后在肝脏水解成具有活性的喹那普利拉,可抑制ACE,阻止血管紧张素I转换为血管紧张素Ⅱ,从而使血管紧张素Ⅱ所介导的血管收缩作用减弱,降低动脉的血管阻力,同时抑制醛固酮的合成,减少醛固酮所产生的水和钠的潴留,使血压下降。
2.本品具有持续24h的长效降压作用。具有降低动脉静脉外周阻力的作用,也能对充血性心力衰竭发挥疗效,是治疗心衰除洋地黄及利尿剂外的主要辅助药。
药代动力学【4】
口服吸收迅速.不受食物影响.吸收率约为60%.在肝脏和消化道内水解成具有更强活性的喹那普利拉 原药达峰时间为1 h.喹耶普利托为2h二者的t1/2分别为(0.89±0.06)h和(2.2±0.2)tl=肝功能减退者原药水解减少,肾功能减退者清除率下降。
作用与用途【4】
本品为无巯基、长效的口服血管紧张素转化酶抑制药.可抑制血管紧张素转化酶.阻止血管紧张素I转化为血管紧张素Ⅱ,从而使血管紧张素Ⅱ所介导的血管收缩作}_H减弱,降低动脉的血管阻力,同时抑制醛吲酮的合成,减少醛同酮所产生的水和钠的潴留.使血压下降本品具有持续24h的长效降压作用,具有降低动,静脉外周阻力的作用,也能对充血性心力衰竭发挥疗效,是除洋地黄及利尿药外治疗心脏衰竭的主要辅助药二适刖于常规药物治疗效果不满意或不良反应较多的高m压及充血性心力衰竭者。
用法用量【3】
本品口服后其吸收不受食物影响。
1.对轻中度高血压推荐起始剂量为每日lOmg,每日1次,如降压效果不满意,可增至每日20~30mg,剂量为每日40rag,每日1次或分2次服用,维持剂量一般为每日lOmg。本品增量时通常要间隔1~2周。对已服用利尿剂的患者,起始剂量应减半。
2.对重度高血压及药物增量后血压下降仍不满意的患者,可加用小剂量的利尿剂(如噻嗪类)或钙拮抗剂。
3.充血性心力衰竭患者在应用利尿剂、强心苷治疗的基础上,推荐本品起始剂量为每日5mg,注意监测患者是否有症状性低血压,剂量可逐渐加量至每次10~20mg,每日2次。
不良反应
该药不良反应的发生率较低,且症状轻。常见的不良反应为干咳、头痛、眩晕、疲劳和感觉异常。干咳与其他ACEI产生机制类似,也是由于体内缓激肽增多刺激支气管而引起,停药后一般可以恢复。其他不良反应 恶心、呕吐、消化不良、腹泻、低m压、皮疹、水肿和瘙痒。偶有血清肌酐及血尿素氮水平升高。肾功能严重减退者可能引起中性粒细胞减少。对于肾动脉狭窄患者,由于降压后可致肾血流量减少,引起肾功能损伤.
药物相互作用【4】
1.与利尿药合用时,因血容量不足或低钠可引起低血压,
2.应避免同时应用保钾利尿药,以免使血钾升高。
3.与洋地黄类药、B受体阻断药、钙拮抗剂等合用不影响相互的药代动力学二
4.与利尿药和(或)B受体阻断药合用能增强降乐作用.
5.本品含碳酸镁,能减少四环素类抗菌药物的吸收,
6.所有血管紧张素转化酶抑制药均会减少锂经肾的排出:
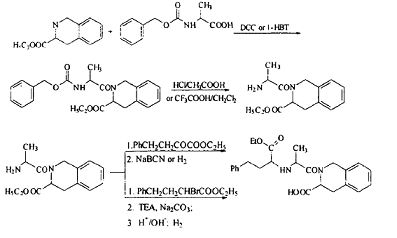
制备【1】
以35一1,2,3,4一四氢异哇琳一3一梭酸乙酷和N一节氧甲酞基丙氨酸为起始原料,以N,N’一二环己基碳二亚胺(DCC)或者l一轻基苯并三吟(1一HBT)为缩合剂,得到缩合产物,脱去保护基后,生成35一N一丙氨酞一1,2,3,4一四氢异喳琳一3一梭酸乙酷,与4一苯基一2一氧代丁酸乙酷反应(脱水、还原),或4一苯基一2一澳代丁酸乙醋在三乙胺(TEA)碱性条件下制备得到哇那普利。
参考文献
[1]祁振海. 第三代血管紧张素转化酶抑制剂盐酸喹那普利的合成研究[D].沈阳药科大学,2002.
[2]丁海英,赵芳,陈伟,许凯,高春环.新型血管紧张素转换酶抑制剂喹那普利的研究进展[J].中国处方药,2005(01):78-80.
[3]杨思进主编,心脑血管药物手册,四川出版集团,2007.5,第49页
[4]魏太星,魏经汉主编,医生专用药物手册,河南科学技术出版社,2015.01,第847页