背景及概述[1][2][3]
巨细胞病毒(Cytomegaoviyns,CMV)是危害性的疱疹病毒之一,人体的感染率高达50%~80%,我国目前成人感染率达到95%以上,一般呈隐性感染,多数感染者无临床症状,但在一定条件下侵袭多个器官和系统可产生严重疾病。病毒可侵入肺、肝、肾、唾液腺、乳腺等其它腺体以及多核白细胞和淋巴细胞,可长期或间歇地自唾液、乳汗、血液、尿液、精液、子宫分泌物多出排除病毒。通过口腔、生殖道、胎盘、输血或器官移植等多种途径传播。
非环状核苷类似物西多福韦是胞嘧啶核苷膦酰基甲醚衍生物,具有广谱抗病毒活性,有效抑制病毒DNA的合成,从而发挥抗病毒作用。1996年,西多福韦注射液被美国食品与药品管理局(FDA)正式批准为艾滋病患者巨细胞病毒(cMv)性视网膜炎的治疗用药,目前已被临床广泛用于CMV性视网膜炎的治疗和其他病毒(疱疹病毒、乳头瘤病毒、腺病毒、多瘤病毒和痘病毒等)的感染。
西多福韦最主要的不良反应为肾毒性,这也是西多福韦限制剂量使用的主要原因。新型广谱抗病毒脂质体Brincidofovir是西多福韦的前体药物,在体内外对DNA病毒和逆转录病毒有强效抗病毒作用,国外正在进行Brincidofovir抗CMV和腺病毒的Ⅲ期临床试验 。Brin.cidofovir化学名为十六烷丙氧基西多福韦,是一种西多福韦和
脂质的联合体,具有可口服、抗病毒作用强和低肾毒性等优点,但对人体一些迅速增殖的细胞如造血干细胞等有一定损害,但尚未获得我国的临床批准。西多福韦能有效治疗疱疹病毒、人乳头瘤病毒和腺病毒等病毒感染以及宫颈上皮内瘤变和牛痘病毒引起的传染性软疣等,选择性西多福韦耐药菌株罕见,且耐药程度较轻,但口服利用度低,不良反应明显,通过对西多福韦进行结构修饰得到其前体药物Brincidofo—viT,抗病毒作用强,肾毒性低,对多种病毒DNA和逆转录病毒有强效抵抗作用,有望成为新型核苷类抗病毒药物。
结构
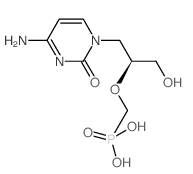
规格[4]
75mg (75mg/mL)。
用法用量[4]
诱导期,5mg/kg,静脉点滴1h以上,每周 1次,疗程2周;维持期,5mg/kg,静脉点滴1h以上,每2周 1次。
应用[4]
用于治疗巨细胞病毒(CMV)感染。
药理作用 [1]
西多福韦是单磷酸胞嘧啶核苷的类似物,需在体内转化为磷酸盐等活性形式,在体内吸收进入细胞后,在酶的作用下转化为活性代谢物,包括单磷酸酯、二磷酸酯和磷酸胆碱盐等。西多福韦的磷酸化作用主要利用宿主细胞内的酶而非病毒的酶 。
西多福韦二磷酸酯通过竞争性抑制胞嘧啶,整合于病毒DNA链,使病毒DNA稳定性降低,从而抑制其延长,发挥抗病毒作用。连续2个西多福韦分子整合人DNA链中会引起DNA链终止延长。Magee WC等 研究由牛痘病毒DNA聚合酶催化的反应和西多福韦在细胞内的活性代谢物西多福韦二磷酸发挥作用的方式。
研究使用不同的引物一模板对和纯化的牛痘病毒DNA聚合酶,观察西多福韦掺人至正在合成的DNA链中并且与模板的鸟嘌呤碱基配对;西多福韦一终结引物也是脱氧单磷酸加成的模板,但2个西多福韦分子同时参加DNA复制的反应产物却不被用于下一轮合成;牛痘病毒聚合酶的校对和核酸外切酶活性可以把西多福韦从引物3 末端切除,但是DNA在倒数第2个3 残基承载着西多福韦,完全可以抵抗核酸外切酶的攻击;通过抑制核酸外切酶校对的活性,西多福韦的错误掺入也可促进病毒复制过程中容易出错的DNA的合成,以降低病毒DNA的稳定性。
药代动力学[1]
滴注西多福韦后,药物的血浆半衰期(t )为2.6 h,西多福韦二磷酸酯和胆碱复合物的t 分别为17.0、48.0 h,较长的半衰期是其在体内抗病毒作用持久的主要原因。西多福韦主要经肾脏代谢,24 h内90%的药物成分经肾小管分泌排泄,具有一定的肾毒性 。西多福韦口服吸收不佳,生物利用<5.3% ,只推荐静脉给药,标准剂量为5 mg/kg,biw或l mg/kg,biw。
西多福韦的生物利用度及药动学研究数据显示,西多福韦单用或与丙磺舒(静脉推注西多福韦前口服丙磺舒2 g,推注后2、8 h各口服1 g)联用,西多福韦的生物利用度均介于5%~20%,血浆清除率分别为(179.0±23.1)、(148.0±38.8)g/L,肾脏清除率分别为(150.0±26.9)、(98.6±27.9) ml/min;西多福二磷酸盐的t 为17 h,胆碱加成物的生物半衰期>48 h,稳态表观分布容积分别是(537±126)、(410±102)L/kg~”。
研究同种异体造血干细胞移植患者血管内西多福韦药动学和安全性的试验显示,6位接受造血干细胞移植且确诊为多瘤病毒家族成员BK病毒或腺病毒感染的患者,给予丙磺舒2 g,3 h后给予3位患者西多福韦5 mg/kg+0.9%氯化钠注射液(Ns)100 ml,通过尿道导管膀胱灌注给药,患者药物耐受性差,调整剂量;给予另3位患者西多福韦2.5 mg/kg+NS 50 ml,ivgt,滴注时间2 h。测得西多福韦表观分布容积、清除率和半衰期分别为19.5 L、5.6 L/h和2.8 h。
不良反应[4]
剂量依赖性的肾毒性、胃肠道反应、皮疹、发热、寒战、粒细胞减少、眼内压减低。偶见或罕见代谢性酸中毒、范可尼 (Fanconi) 综合征、眼色素层膜、乏力。
注意事项[4]
1. 孕妇及哺乳期妇女禁用。
2. 对本品或丙磺舒过敏者禁用。
3. 血肌酐值>1.5mg/dL或血肌酐值超过正常范围0.5mg/dL或尿蛋白≥100mg/dL或CrCl<55mL/min者禁用。
4. 用药前1周避免使用其他肾毒性药物。
5. 静脉给药前3h口服丙磺舒2g,给药后2h和8h口服丙磺舒各1g,用药前需要静滴1000mL生理盐水水化。
药物相互作用[2]
西多福韦与复方新诺明、双脱氧肌苷、氟康唑和氨基糖甙类抗生素有明显的药代动力学相互作用。HIV 患者用齐多夫定和西多福韦联合用药不影响齐多夫定的AUC。在输注西多福韦前3小时注射1 g丙磺舒或在输注西多福韦后2和8小时注射0.5 g丙磺舒及1升的生盐水不影响西多福韦的药代动力学性质,若丙磺舒的剂量加倍,并与生理盐水一起注射,能使西多福韦的血浓度提高2倍,并可降低其肾毒性,这可能是通过阻滞西多福韦在肾小管的排泄,降低其清除率至接近肾小球过滤的水平。用西多福韦治疗,同时口服丙磺舒也有类似的作用。
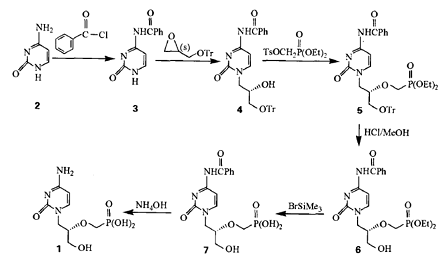
制备[3][5]
方法1:以胞嘧啶为起始原料(2),经过苯甲酰化得到氨基保护胞嘧啶(3),与(S)一三苯甲氧基甲基环氧乙醇缩合得到(s)一N4一苯甲酰基一N 一【(2一羟基一3一三苯氧基)丙基】胞嘧啶(4)、再与对甲苯磺酰氧甲基亚磷酸二乙酯侧链缩合引入磷脂基得到西多福韦的多保护物(S)一N‘一苯甲酰基一N 一【(2一磷酸二乙酯甲氧基一3一三苯甲氧基)丙基】胞嘧啶(5)。
中间体5中的保护基分别采用不同方法脱去,首先用氯化氢催化醇交换法脱去三苯甲基游离出丙基侧链羟基(6)、再用三甲基溴化硅脱去磷脂乙基游离出磷酸基(7)、最后在氨水作用下脱去苯甲酰基得到目的产物西多福韦(1)。
合成中所用原料胞嘧啶和主要试剂(R)一缩水甘油国内均有生产,磷脂侧链对甲苯磺酰氧甲基亚磷酸二乙酯采用文献b 报道方法合成得到。采用该路线合成西多福韦的总收率为23.1% ,结构经 H—NMR和MS确证,产品纯度在99% 以上。本法合成西多福韦原料易得,操作简便,易于工业化生产。
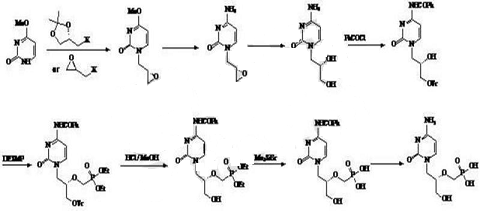
方法2:从原料(S)-羟甲基环氧乙烷出发,合成(S)-1-R-2,3-环氧丙烷(R为烃基),经醚化,酰化,縮合,脱保护基,水解,酸化等反应制备西多福韦。
1)在0~50℃环境温度和有机胺碱、催化剂和溶剂存在的条件下,将手性羟甲基环氧乙烷和卤代烃RX发生反应,得到;所述手性羟甲基环氧乙烷、催化剂、卤代烃RX和有机胺碱的投料摩尔比为1~5︰0.01~0.1︰1︰1~5;所述卤代烃RX中R为苄基或C1-4的烷基或三苯基甲基及其衍生物或对甲苯磺酰基,X为Cl,Br,I;所述有机胺碱为NEt3或N,N-二甲基苯胺或N,N-二乙基苯胺;所述溶剂为CH2Cl2、CHCl3、苯、甲苯或二甲苯中的任意至少一种;所述催化剂为4-二甲氨基吡啶;
2)于50~140℃温度条件下,将胞嘧啶在反应溶剂中,经碱脱质子后,再与发生亲核取代反应,减压蒸馏出溶剂,经洗涤溶剂洗涤后,干燥得到;所述、碱和胞嘧啶的投料摩尔比为1︰0.8~1.0︰1~1.3;所述的碱为t-BuOK或t-BuONa或t-BuOLi或K2CO3或Cs2CO3或NaH或LiH;所述反应溶剂为N,N-二甲基甲酰胺、N,N-二乙基甲酰胺、N,N-二甲基乙酰胺、二氧六环或二甲亚砜中的至少一种;所述洗涤溶剂为以甲酸甲酯、甲酸乙酯、乙酸甲酯、乙酸乙酯、丙酸乙酯、二氯甲烷或氯仿中的至少任意一种;
3)在20~120℃温度下,于溶剂中,将经叔丁醇镁脱质子后,再与对甲苯磺酰氧甲基亚膦酸二乙酯(DESMP)发生亲核取代反应,采用酸中和后,经减压蒸馏出溶剂,再经洗涤、过滤、浓缩得到;所述和叔丁醇镁的投料摩尔比为1︰0.5~2;所述溶剂为N,N-二甲基甲酰胺、N,N-二乙基甲酰胺、N,N-二甲基乙酰胺、二氧六环或二甲亚砜中的至少任意一种;所述中和用的酸为甲酸或醋酸或丙酸或苯甲酸或对甲苯磺酸或对甲苯磺酸含水结晶物;所述洗涤用的溶剂为甲酸甲酯、甲酸乙酯、乙酸甲酯、乙酸乙酯、丙酸乙酯、四氢呋喃、二氯甲烷、氯仿中的至少任意一种;
4)在70~120℃温度条件下,将在醋酸的作用下脱保护反应后,经减压蒸馏、洗涤、过滤浓缩得到;所述醋酸的体积百分比浓度为50~100%;所述洗涤用的溶剂为乙腈、乙酸乙酯、二氯甲烷、氯仿中的至少任意一种;
5)在20-120 ℃温度条件下,将与质量百分比为10-80%的HX水溶液反应后,调节pH值至2-4,析出固体,再将固体经醇或水重结晶得到西多福韦;
主要参考资料
[1] 房舒舒, 张帅, 曹国颖. 核苷类抗病毒药物西多福韦的研究进展[J]. 中国药房, 2016, 27(21): 3022-3024.
[2] 陈历胜. 抗巨细胞病毒新药—西多福韦[J]. 国外医药: 合成药. 生化药. 制剂分册, 1998, 19(1): 28-31.
[3] 易红, 李卓荣. 抗病毒药物西多福韦的合成研究[J]. 中国抗生素杂志, 2006, 31(7): 412-413.
[4] 协和抗感染手册
[5] CN201110260086.8 抗病毒药物西多福韦的一种合成方法