背景技术
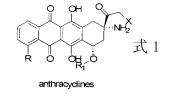
文献(Synthetic Communications,2004,34(17),3047-3059)披露 5,8-二甲氧 基-2-萘满酮是制备蒽环霉素类(anthracyclines,参见式1)抗癌药物的重要中间体。欧洲 专利EP0343830披露5,8- 二甲氧基-2-萘满酮还是制备血液中复合胺再吸收抑制剂2-哌 嗪四氢萘的重要中间体。欧洲专利EP00343830披露5,8-二甲氧基-2-萘满酮是制备2-胺 基萘类抗细菌和抗肿瘤药物的重要中间体。
现有技术中合成5,8- 二甲氧基-2-萘满酮的方法主要有以下几种:
一 是文献(J.Am· Chem. Soc.,1952,74 (5) ,5321-5324)披露的以 2-乙氧基-1, 3_丁二烯和1,4_苯醌为起始原料经过Diels-Aldel反应、烯醇化、酚羟基的保护、双键的迁 移、脱乙基等步骤制备5,8_ 二甲氧基-2-萘满酮的方法,参见式2。该方法存在两个缺陷: 1。其起始原料2-乙氧基-1,3-丁二烯难以制备,收率很低;2。整个的合成路线较长,总收 率也较低。

另一个方法是文献(Synthetic Communications,2004,34 (17),3047-3059)披露 的以丁二烯和对苯醌为起始原料经过Diels-Aldel反应、烯醇醚化、环氧化、开环氧成酮等 步骤合成5,8-二甲氧基-2-萘满酮的方法(参见式3)。该方法中环氧开环氧成酮反应收 率很低,而且不易纯化。

该文献还提供了另外一种方法:2_氯-1,3-丁二烯与1,4_苯醌经过Diels-Aldel 反应、烯醇醚化、水解等步骤(参见式4)。该方法中起始原料2-氯-1,3-丁二烯难以制备, 而且最后水解的收率很低。

式4
欧洲专利EP00343830和德国专利DE3720129还提供了另外一种方法:1,4_ 二甲 氧基萘经过Na/EtOH还原后得到1,4- 二甲氧基-5,8- 二氢萘,然后用间氯过氧苯甲酸惊醒 环氧化,再用LiAlH4还原开环氧,用PCC氧化醇成酮,即得到5,8- 二甲氧基-2-萘满酮(参 见式5)。该合成方法也存在两个不足:1。开环氧需要LiAlH4,在工业生产上很危险,也不 易操作;2。PDC氧化的收率很低。

式 5
另外,文献(Liebigs Annalen der Chemie, (2) ,435-7 ; 1985)则提供了从5,8_二 甲氧基-1-萘满酮经过还原、脱水、环氧化合开环氧等步骤制备5,8- 二甲氧基-2-萘满酮 的方法(参见式6)。该方法所需的原料5,8_ 二甲氧基-1-萘满酮难以制备。同时,

发明内容
本发明提供一种可克服现有技术不足合成5,8_ 二甲氧基-2-萘满酮的方法。
本发明的方法以对苯醌和1,3-丁二烯为起始原料经过Diels-Aldel反应生成4a, 5,8,8a-四氢-1,4-萘醌。然后通过烯醇化、醚化将4a, 5,8,8a-四氢-1,4-萘醌转化为5, 8- 二甲氧基-1,4- 二氢萘。将5,8- 二甲氧基-1,4- 二氢萘在碱催化下转化为5,8- 二甲氧 基-1,2- 二氢萘后,然后进行环氧化得到5,8- 二甲氧基-1,2-环氧-3,4- 二氢萘,在酸催 化下环氧开环得到5,8_ 二甲氧基-2-萘满酮。参见式7:

式 7
本发明的一个具体实施例中,其合成方法是:
a.将对苯醌溶于醋酸中,搅拌下,向反应液中通入丁二烯气体至过量,继续搅拌至 反应完全后,将反应液倒入冰水中,搅拌,过滤,滤饼用水洗后晾干得4a,5,8,8a-四氢-1,4-萘醌。
b.将上述所得4a,5,8,8a-四氢-1,4-萘醌悬浮于水中,加入2至10倍摩尔量的 ΚOΗ,加热回流搅拌反应3小时后,向反应液中缓慢滴入2至5倍摩尔量硫酸二甲酯,加完后 继续反应至原料消失,将反应液冷却至室温后,倒入冰水中搅拌,过滤,滤饼水洗,晾干或者 真空干燥得到5,8- 二甲氧基-1,4- 二氢萘。
c.将上述所得5,8_ 二甲氧基-1,4-二氢萘溶于DMSO中,向溶液中加入0.05至 1. 0倍摩尔量的叔丁醇钾或者叔丁醇钠,反应至原料消失,将反应液倒入冰水中,搅拌,过 滤,水洗,滤饼晾干或者真空干燥得到5,8- 二甲氧基-1,2- 二氢萘。
d.将上述所得5,8_ 二甲氧基-1,2-二氢萘溶于二氯乙烷中,向其中加入1.5至5 倍摩尔量NaHCO3饱和水溶液,然后缓慢向其中滴入1. 0至1. 5倍摩尔量间氯过氧化苯甲酸 的饱和二氯乙烷溶液。室温反应至原料消失,二氯甲烷萃取,合并有机相后,用亚硫酸钠、亚 硫酸氢钠或者硫代硫酸钠水溶液洗去过量的过氧化物后,浓缩得到5,8- 二甲氧基-1,2-环 氧-3,4-二氢萘的粗产物。
e.将上述5,8-二甲氧基-1,2-环氧-3,4_ 二氢萘粗产物溶于乙醇中,向其中加入 盐酸,加热回流至原料消失,减压蒸去溶剂,残余物经过萃取、结晶纯化或者减压蒸馏、重结 晶纯化得目标化合物5,8-二甲氧基-2-萘满酮。
本发明所提供的方法以对苯醌和1,3-丁二烯为起始原料,经过Diels-Aldel反应、烯醇醚化、双键迁移、环氧化和开环氧五步反应,以32〜36%的总收率得到5,8_二甲氧 基-2-萘满酮。相关的试验表明,本发明所用环氧化试剂可以为间氯过氧化苯甲酸、过氧化乙酸、0Χ0ΝΕ、David’ s试剂等。环氧化反应可以在碱存在下反应也可以在无碱的条件下反 应,这里的碱包括NaHCO3, Na2CO3, K2CO3, NaOH,等。本发明所用的开环氧的试剂可以为质子 酸或者Lewis酸,质子酸包括盐酸、硫酸、对甲苯磺酸,等,Lewis酸包括ZnI2, BF3 · Et2O,等。
本发明具有以下优点:1。本发明中所涉及的原料、试剂都十分易得,价格便宜;2。 本发明提供的方法中,所生成的中间体都十分容易纯化;3。本方法中的反应条件温和,适合 于工业生产。
具体实施方式
以下提供本发明的实施例。
将对苯醌25克溶于45毫升醋酸中,搅拌下,于10±5摄氏度下,在1小时内向反 应液中通入38克丁二烯气体,通完气体后,继续搅拌至反应10小时后,将反应液倒入冰水 250毫升中,用200毫升乙醚萃取三次,合并有机相依次用饱和碳酸氢钠洗,饱和食盐水洗, 无水硫酸钠干燥,过滤蒸干后,残余物用硅胶柱层析纯化得4a,5,8,8a-四氢-1,4-萘醌30 克,收率80%。
将25. 3克4a,5,8,8a-四氢-1,4-萘醌悬浮于120克水中,加入33. 6克ΚOΗ,加热 回流搅拌反应3小时后,向反应液中缓慢滴入57克硫酸二甲酯,加完后继续加热回流反应 5小时后,将反应液冷却至室温后,用100毫升正己烷萃取三次,合并有机相依次用水洗,饱 和食盐水洗,无水硫酸钠干燥,过滤蒸干后,残余物用硅胶柱层析纯化得5,8- 二甲氧基-1, 4-二氢萘25. 1克,收率88%。 将19克5,8- 二甲氧基-1,4- 二氢萘溶于40毫升DMSO中,向溶液中加入3. 4克 叔丁醇钾,室温反应6小时后,冷却至室温,将反应液倒入2升冰水中,用100毫升正己烷萃 取三次,合并有机相依次用水洗,饱和食盐水洗,无水硫酸钠干燥,过滤蒸干后,残余物用硅 胶柱层析纯化得5,8- 二甲氧基-1,2- 二氢萘18克,收率95 %。
将15. 2克5,8-二甲氧基-1,2-二氢萘溶于30毫升二氯甲烷中,向其中加入34 克NaHCO3的饱和水溶液,然后向其中加入20. 3克85%的间氯过氧苯甲酸的200毫升二氯 甲烷溶液。室温反应搅拌反应1小时后,静置分层,分液后,水相用100毫升二氯甲烷萃取 三次,合并有机相用硫代硫酸钠水溶液洗,饱和食盐水洗,无水硫酸钠干燥,过滤蒸干后,残 余物用过胶柱层析纯化得5,8- 二甲氧基-1,2-环氧-3,4- 二氢萘10. 3克,收率63%。
将8. 3克5,8- 二甲氧基-1,2-环氧_3,4- 二氢萘溶于60毫升甲苯中,向其中加 入无水2. 6克ZnI2,加热回流1小时后,加20毫升水淬灭反应,然后静置分液,水相用50毫 升二氯甲烷萃取三次,合并有机相用20毫升饱和NaHCO3水溶液洗,20毫升饱和食盐水洗, 无水硫酸钠干燥,过滤蒸干后,残余物用乙醇重结晶得5,8- 二甲氧基-2-萘满酮6. 2克,收 率 75%。