|
ChemicalBook Optimization Suppliers |
|
| 融点 | 214 °C | | 比旋光度 | D24 +153° (c = 0.5 in water) | | 沸点 | 386.09°C (rough estimate) | | 比重(密度) | 1.3686 (rough estimate) | | 屈折率 | 1.5100 (estimate) | | 貯蔵温度 | 2-8°C | | 溶解性 | H2O: 50 mg/mL, clear, colorless | | 外見 | crystalline | | 酸解離定数(Pka) | pKa 4.3 (Uncertain) | | 色 | Prisms from EtOH (aq) | | 水溶解度 | almost transparency | | 極大吸収波長 (λmax) | 272nm(H2O)(lit.) | | Merck | 14,2784 | | BRN | 89175 | | 安定性: | Stable for 1 year as supplied from date of purchase. Solutions in DMSO or water may be stored at -20°C for up to 1 month. | | InChIKey | UHDGCWIWMRVCDJ-STUHELBRSA-N | | LogP | -1.808 (est) | | CAS データベース | 147-94-4(CAS DataBase Reference) | | EPAの化学物質情報 | Cytarabine (147-94-4) |
| | シタラビン Usage And Synthesis |
| 外観 | 白色~ほとんど白色粉末~結晶 | | 用途 | シトシン系代謝拮抗物質です。
DNA ポリメラーゼⅠ阻害作用を示します。 | | 効能 | 抗悪性腫瘍薬, 代謝拮抗薬, 抗ウイルス薬, DNAポリメラーゼ阻害薬 | | 商品名 | キロサイド (日本新薬); キロサイド (日本新薬) | | 説明 | Cytarabine (147-94-4) is a nucleoside analog that interferes with DNA synthesis and transcription, particularly in tumor cells.1,2 Cellular enzymes convert it to its nucleotide form, Ara-CTP, which disrupts DNA and RNA synthesis when polymerases attempt to incorporate it.3,4 For the same reasons, it is also employed as an antiviral.5 It inhibits proliferation of a variety of leukemic cell lines (IC50s 16-72 nM), but is used primarily against acute myeloid leukemia (AML).6 | | 化学的特性 | A white or almost white, crystalline powder, freely soluble in water, very slightly soluble in alcohol and in methylene chloride. | | Originator | Cytosar,Upjohn,US,1969 | | 使用 | antineoplastic, antiviral, antimetabolite | | 使用 | Cytarabine USP (Cytosar)is used to treat Acute granulocytic leukemia (adults); acute lymphocytic leukemia (children); Hodgkin’s disease | | 使用 | Used as an antineoplastic and antiviral. A selective inhibitro of DNA synthesis. Does not inhibit RNA synthesis | | 定義 | ChEBI: A pyrimidine nucleoside in which cytosine is attached to D-arabinofuranose via a beta-N1-glycosidic bond. | | 適応症 | Cytarabine (cytosine arabinoside, ara-C, Cytosar-U) is
an analogue of the pyrimidine nucleosides cytidine and
deoxycytidine. It is one of the most active agents available
for the treatment of acute myelogenous leukemia.
Cytarabine kills cells in the S-phase of the cycle by competitively
inhibiting DNA polymerase. The drug must
first be activated by pyrimidine nucleoside kinases to
the triphosphate nucleotide ara-cytosine triphosphate
(ara-CTP). The susceptibility of tumor cells to cytarabine
is thought to be a reflection of their ability to activate
the drug more rapidly (by kinases) than to inactivate
it (by deaminases). | | Manufacturing Process | (A) Preparation of 1- (2,3,5-Tri-O-Acetyl-β-D-Arabinofuranosyl)-4-Thiouracil: A
mixture of 1.85 g (5.0 mmol) of 1-(2,3,5-tri-O-acetyl-β-arabinofuranosyl)
uracil, 1.23 g (5.55 mmol) of phosphorus pentasulfide, and 30 ml of pyridine
was heated under gentle reflux for 2.5 hours with exclusion of moisture. The
reaction mixture was cooled, and the supernatant solution was transferred by
means of a pipette into a mixture of crushed ice and water. The reaction flask
was washed twice with pyridine, and these washings were added to the icewater mixture. This mixture was kept at about 25°C until the ice had melted,
and was then stored at 0°C for one hour. A pale yellow precipitate that formed
was collected on a filter, washed with ice-water, and dried in air.
This material was triturated with chloroform, and the chloroform mixture was
filtered. A small amount of undissolved material collected on the filter and it was washed with chloroform. The chloroform solution (filtrate plus washings)
was washed three times with ice-water, twice with ice-cold 3 N sulfuric acid,
twice with ice-cold saturated aqueous sodium bicarbonate solution, twice with
ice-water, and then dried over anhydrous sodium sulfate. The chloroform was
removed under reduced pressure at a bath temperature of about 40°C,
leaving a yellow, somewhat gummy residue. This yellow residue was dissolved
in absolute methanol which was then evaporated at reduced pressure at about
40°C, and the residue was then held for 2 hours at 0.5 to 2.0 mm pressure
and a bath temperature of about 50°C. There was thus obtained 1.69 g of 1-
(2,3,5-tri-O-acetyl-β-D-arabinofuranosyl)-4-thiouracil.
(B) Preparation of 1-β-D-Arabinofuranosylcytosine: In a glass liner, a mixture
of 1.16 g (3.0 mmol) of 1-(2,3,5-tri-O-acetyl-β-D-arabinofuranosyl)-4-
thiouracil prepared in (A) and about 60 ml of absolute methanol which had
been saturated with anhydrous ammonia at 0°C was heated in a steel bomb
at 98° to 105°C for 35 hours. After cooling to about 25°C and venting the
bomb, the dark solution was filtered into a round-bottom flask. The methanol
and excess ammonia were then removed under reduced pressure at about
25°C. The residual syrup was dissolved in absolute methanol, and the
methanol was removed under reduced pressure at a bath temperature of
about 40°C. This procedure of dissolving in absolute methanol and removing
the solvent was repeated, and the residue was held under reduced pressure at
a bath temperature of 45°C for 12 hours.
The resulting semisolid was triturated thoroughly with absolute methanol, and
the resulting suspension was chilled at 0°C. A pale tan solid that separated
was collected on a filter and washed repeatedly with methanol. After washing
with anhydrous ether, there was obtained 430 mg of 1-β-Darabinofuranosylcytosine.
(C) Preparation of 1-β-D-Arabinofuranosylcytosine Hydrochloride: The absolute
methanolic filtrate obtained after triturating and filtering the 1-β-Darabinofuranosylcytosine in (B) above was warmed and stirred with
decolorizing charcoal. The mixture was filtered through a bed of filter aid, and
the filter bed was washed repeatedly with absolute methanol. The combined
filtrate and washings were pale yellow. The solution was diluted to faint
cloudiness with anhydrous ether, and an excess of anhydrous hydrogen
chloride was introduced. Crystallization began at about 25°C and further
crystallization was induced by chilling at 0°C for 14 hours. The crystalline
product was collected on a filter, washed with anhydrous ether, and dried in
air. There was thus obtained 180 mg of pale yellow 1-β-Darabinofuranosylcytosine hydrochloride melting at 186° to 189°C.
The pale yellow product was dissolved in warm, absolute methanol, and the
solution after mixing with decolorizing charcoal was filtered through a bed of
filter aid. The filter bed was washed with warm absolute methanol, and the
combined methanolic filtrate and washings were warmed and diluted with
anhydrous ether to incipient crystallization. The methanol-ether mixture was
kept at about 25°C for about 1 hour and then chilled, first at 0°C, and then at
-20°C. The resulting colorless needles were collected on a filter, washed with
anhydrous ether, and dried at 85°C, yielding 100 mg of 1-β-Darabinofuranosylcytosine hydrochloride having a melting point of 186° to
188°C. | | brand name | Cytosar-U (Sicor); Depocyt (Skyepharma). | | Therapeutic Function | Cancer chemotherapy | | 一般的な説明 | Colorless crystals. Used as an antiviral agent. | | 一般的な説明 | Cytarabine is a pyrimidine nucleoside drug that is related toidoxuridine. This agent is primarily used as an anticanceragent for Burkitt lymphoma and myeloid and lymphaticleukemias. Cytarabine blocks the cellular utilization of deoxycytidine,hence inhibiting the replication of viral DNA.Before it becomes active, the drug is converted to monophosphates,diphosphates, and triphosphates, which block DNApolymerase and the C-2 reductase that converts cytidinediphosphate into the deoxy derivative.
The antiviral use of cytarabine is in the treatment of herpeszoster (shingles), herpetic keratitis, and viral infectionsthat resist idoxuridine. Cytarabine is usually administeredtopically. Toxicity occurs on bone marrow, the gastrointestinal(GI) tract, and the kidneys. | | 一般的な説明 | The drug is available in 100-, 500-, 1,000-, and 2,000-mgmultidose vials for IV use. Cytarabine is used in the treatmentof acute myelogenous leukemia and CML. This drugis a deoxycytidine analog originally isolated from thesponge Cryptothethya crypta. It is active following intracellularactivation to the nucleotide metabolite ara-CTP. Theresulting ara-CTP is incorporated into DNA resulting inchain termination and inhibition of DNA synthesis andfunction. Resistance can occur because of decreased activationor transport and increased catabolic breakdown.Metabolic breakdown within the GI tract leads to poorbioavailability. The drug distributes rapidly into tissues andtotal body water with cerebrospinal fluid (CSF) levelsreaching 20% to 40% of those in plasma. Cytidine deaminaseis the primary catabolic enzyme involved in the inactivationof cytarabine. Drug interactions include antagonismof the effects of gentamicin, decreasing the oral bioavailabilityof digoxin, as well as enhancing the cytotoxicity ofvarious alkylating agents, cisplatin, and ionizing radiation.
Toxicities include myelosuppression, leukopenia andthrombocytopenia, nausea and vomiting anorexia, diarrhea,and mucositis. Neurotoxicity is usually expressed as ataxia,lethargy, and confusion. An allergic reaction often describedin pediatric patients includes fever, myalgia, malaise, bonepain, skin rash, conjunctivitis, and chest pain.Pretreatment with methotrexate enhances the formation ofara-CTP metabolites resulting in enhanced cytotoxicity. | | 空気と水の反応 | Water soluble. | | 健康ハザード | ACUTE/CHRONIC HAZARDS: Very toxic. Hazardous decomposition products. May cause irritation on contact. Teratogen. Mutagen. Central nervous system effects. | | Biochem/physiol Actions | Ara-C incorporates into DNA and inhibits DNA replication by forming cleavage complexes with topoisomerase I resulting in DNA fragmentation; does not inhibit RNA synthesis. Anti-leukemia agent. | | 作用機序 | Cytarabine is rapidly metabolized in the liver, kidney,
intestinal mucosa, and red blood cells and has a
half-life in plasma of only 10 minutes after intravenous
bolus injection. The major metabolite, uracil arabinoside
(ara-U), can be detected in the blood shortly after
cytarabine administration. About 80% of a given
dose is excreted in the urine within 24 hours, with less
than 10% appearing as cytarabine; the remainder is
ara-U.When the drug is given by continuous infusion,
cytarabine levels in CSF approach 40% of those in
plasma. | | 臨床応用 | Cytarabine is used in the chemotherapy of acute
myelogenous leukemia, usually in combination with an
anthracycline agent, thioguanine, or both. It is less useful
in acute lymphoblastic leukemia and the lymphomas
and has no known activity against other tumors. It has
been used intrathecally in the treatment of meningeal
leukemias and lymphomas as an alternative to methotrexate. | | 安全性プロファイル | Moderate to low toxicity byingestion. Human systemic effects: allergic dermatitis,ataxia, blood changes, central nervous system effectsconjunctive irritation, degenerative brain changes, hearingacuity change, lachrymation, peripheral nervefasciculati | | 合成 | Cytarabine, 4-amino-1-|?-arabinofuranosyl-2(1H)pyrimidone (30.1.3.8), is
made from 1-|?-D-arabinofuranosyluracil by preliminary acylation of the hydroxyl group,
forming a triacetyl derivative (30.1.3.6), and subsequent replacement of the carbonyl group at
position 4 of the pyrimidine ring with a thiocarbonyl group using phosphorous pentachloride,
and finally replacing the mercapto group of 30.1.3.7 with an amino group using ammonia and
simultaneous hydrolysis of the acetyl-substituted groups, giving cytarabine (30.1.3.8). 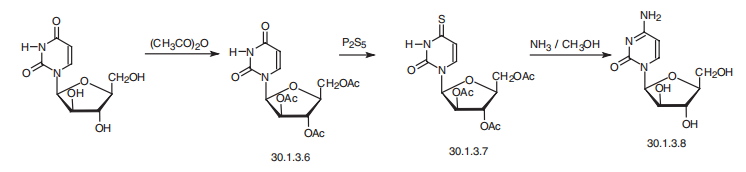
| | Veterinary Drugs and Treatments | In veterinary medicine, cytarabine is used primarily in small animals
as an antineoplastic
agent for lymphoreticular neoplasms,
myeloproliferative disease (leukemias), and CNS lymphoma. Refer
to the Dosages below or the Protocols (in the appendix), for more
information. | | 薬物相互作用 | Potentially hazardous interactions with other drugs
Antipsychotics: avoid with clozapine, increased risk
of agranulocytosis. | | 代謝 | Cytarabine is converted by phosphorylation to an active
form, which is rapidly deaminated, mainly in the liver and
the kidneys, by cytidine deaminase to inactive 1-β-d�arabinofuranosyluracil (uracil arabinoside, ara-U).
Approximately 80% of an intravenous dose is excreted
in the urine within 24 hours, mostly as the inactive
metabolite with about 10% as unchanged cytarabine. A
small amount is excreted in the bile. | | 貯蔵 | Store at +4°C | | 純化方法 | Purify cytarabin by recrystallisation from aqueous EtOH or a large volume of H2O (it solubility at ~20o is 5%). It has max 212 and 279nm at pH 2 and 272nm at pH 12. It is an acute leukaemic agent. [Walwick et al. Proc Chem Soc (London) 84 1959, Beilstein 25 III/IV 3669.] | | 参考文献 | Derissen and Beijnen (2020), Intracellular Pharmacokinetics of Pyrimidine Analogues used in Oncology and the Correlation with Drug Action; Clin. Pharmacokinet., 59 1521
Z Li et al. (2017), Exploring the Antitumor Mechanism of High-Dose Cytarabine through the Metabolic Perturbations of Ribonucleotide and Deoxyribonucleotide in Human Promyelocytic Leukemia HL-60 Cells; Molecules, 22 E499
Zhang & Kiechle (2004), Cytosine Arabinoside Substitution Decreases Transcription Factor-DNA Binding Element Complex Formation; Arch. Pathol. Lab. Med., 128 1364
Renis (1973), Antiviral Activity of Cytarabine in Herpesvirus–Infected Rats; Antimicrob. Agents Chemother., 4 439
Qin et al. (2007), Effect of Cytarabine and Decitabine in Combination in Human Leukemic Cell Lines; Clin. Cancer Res., 13 4225
Walter et al. (2020), Optimal Dosing of Cytarabine in Induction and Post-Remission Therapy of Acute Myeloid Leukemia; Leukemia, 35 295 |
|