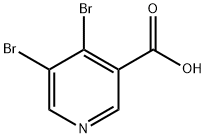
4,5-dibroMonicotinic acid synthesis
- Product Name:4,5-dibroMonicotinic acid
- CAS Number:1009334-28-4
- Molecular formula:C6H3Br2NO2
- Molecular Weight:280.9
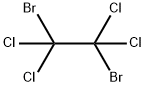
630-25-1
1009334-28-4
5-Bromonicotinic acid (25.25 g, 125 mmol) was dissolved in anhydrous tetrahydrofuran (500 mL) under nitrogen protection, stirred and cooled to -70 °C. Lithium diisopropylammonium (1.8 M, 144 mL, 260 mmol) was added slowly and dropwise over 1 hour. After the dropwise addition, the reaction mixture was continued to stir at -55 °C for 2.5 h, followed by cooling to -70 °C again. 1,2-Dibromotetrachloroethane (50 g, 154.5 mmol) was added in batches over 30 minutes. Once the addition was complete, stirring was continued for 30 minutes, followed by a slow warming to -20°C over 2 hours. After careful addition of water (150 mL), the organic solvent was removed by distillation under reduced pressure. The residue was diluted with water (500 mL) and washed with ethyl acetate. Subsequently, the aqueous phase was acidified with hydrochloric acid to pH 3.00. The precipitated solid product was collected by filtration and dried under vacuum at 60 °C to give 4,5-dibromonicotinic acid (14.2 g). The filtrate was extracted with ethyl acetate, the organic phases were combined, washed with water, dried over anhydrous magnesium sulfate, filtered and concentrated under reduced pressure to give additional 4,5-dibromonicotinic acid (18.6 g, total yield 32.8 g, 93% yield). The product was characterized by 1H NMR (DMSO-d6, 400 MHz): δ 8.92 (s, 1H), 8.73 (s, 1H).
630-25-1
171 suppliers
$5.00/1g
1009334-28-4
24 suppliers
$121.00/100mg
Yield:1009334-28-4 93%
Reaction Conditions:
Stage #1: 5-bromo-3-pyridinecarboxylic acidwith lithium diisopropyl amide in tetrahydrofuran at -70 - -55; for 3.5 h;
Stage #2: 1,2-dibromo-1,1,2,2-tetrachloroethane in tetrahydrofuran at -70 - -20; for 2.5 h;
Steps:
1
5-Bromonicotinic acid (25.25 g, 125 mmol) was stirred as a solution in dry tetrahydrofuran (500 ml) under nitrogen and cooled to -7O0C. The resultant mixture was treated dropwise over 1 hour with lithium diisopropylamide (1.8 M, 144 ml, 260 mmol). After the addition was complete, the solution was stirred for 2.5 hours at - 550C then cooled to -7O0C and treated portionwise over 30 minutes with 1,2-dibromotetrachloroethane (50 g, 154.5 mmol). After stirring for 30 minutes the mixture was allowed to warm to -2O0C over 2 hours before the cautious addition water (150 ml). The organic solvent was then removed in vacuo and the residue diluted with water (500 ml), then washed with ethyl acetate before acidifying the aqueous layer to pH 3.00 with c. HCl. The precipitated product was collected by filtration and dried at 6O0C in vacuo to give the title compound (14.2g). The filtrate was extracted with ethyl acetate, the extract washed with water, dried (MgSO4), filtered and concentrated in vacuo to give further title compound (18.6g, total yield 32.8 g, 93%). 1H NMR (DMSO-d6, 400MHz) 8.92 (s, IH), 8.73 (s, IH).
References:
WO2008/24725,2008,A1 Location in patent:Page/Page column 93
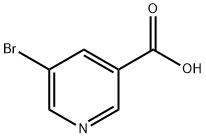
20826-04-4
553 suppliers
$5.00/1g
1009334-28-4
24 suppliers
$121.00/100mg