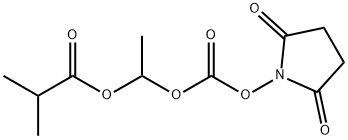
Propanoic acid, 2-Methyl-, 1-[[[(2,5-dioxo-1-pyrrolidinyl)oxy]carbonyl]oxy]ethyl ester synthesis
- Product Name:Propanoic acid, 2-Methyl-, 1-[[[(2,5-dioxo-1-pyrrolidinyl)oxy]carbonyl]oxy]ethyl ester
- CAS Number:860035-10-5
- Molecular formula:C11H15NO7
- Molecular Weight:273.24
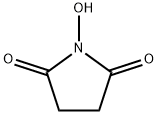
6066-82-6
787 suppliers
$5.00/10g
![1-{[(methylsulphanyl)carbonyl]oxy}ethyl 2-methylpropanoate](/CAS/20200331/GIF/860035-07-0.gif)
860035-07-0
2 suppliers
inquiry
860035-10-5
17 suppliers
$448.00/1g
Yield:860035-10-5 82%
Reaction Conditions:
with magnesium monoperoxyphthalate hexahydrate in dichloromethane at 20 - 25; for 5 h;Product distribution / selectivity;
Steps:
5.10.10
5.10 Example 10: f (l-Isobutanoyloxyethoxy) carbonyloxyl Succinimide (10) To a solution of compound (6) (1 g, 4.8 mmol) in CH2C12 (10 mL) was added N-hydroxysuccinimide (1.1 g, 9.5 mmol) and the reaction mixture cooled to 0°C. A solution of 32% (v/v) peracetic acid in acetic acid (3.4 mL, 1.1 g, 14.4 mmol) was added dropwise over a period of 10 min, then the solution allowed to stir at room temperature for 3 h. The reaction mixture was diluted with ether (50 mL) and washed with water (2 x 10 mL), saturated sodium bicarbonate solution (10 mL) and brine (10 mL), then dried over anhydrous sodium sulfate, filtered and concentrated in vacuo to give the title compound (10) as a as colorless oil (1 g, 77%). After trituration with hexane (20 mL) the product solidified to a white solid. m. p: 50-54°C. IH NMR (CDC13, 400 MHz) : 8 1. 17 (d, J=6.8 Hz, 6H), 1.56 (d, J=5. 6 Hz, 3H), 2.55 (m, 1H), 2.82 (s, 4H), 6.80 (q, J= 5.2 Hz, 1H). MS (ESI) mlz 296.4 (M+Na) +. In an alternative synthesis of (10), N-hydroxysuccinimide (558 mg, 4.8 mmol) was added to a solution of compound (6) (500 mg, 2.4 mmol) in CH2C12 (10 mL) and the reaction mixture cooled to 0°C. m-Chloroperbenzoic acid (1.62 g, 7.2 mmol, commercial grade: 77% in water) was added over a period of 10 min and the mixture allowed to stir at room temperature for 16 h. The reaction mixture was diluted with ether (50 mL) and washed with water (2 x 10 mL), saturated sodium bicarbonate solution (10 mL) and brine (10 mL). The organic layer was dried over anhydrous sodium sulfate, filtered and concentrated in vacuo to give the title compound (10) together with m-chlorobenzoic acid. The crude product mixture was purified by column chromatography on silica gel, eluting with 4: 6 EtOAc: hexane. Residual m-chlorobenzoic acid was removed by repeated crystallizations from a mixture of tert-butyl methyl ether and hexane, resulting in analytically pure product (66 mg, 10%). In an alternative synthesis of (10), a 500-mL, three-neck flask equipped with a mechanical stirrer, teflon-coated thermocouple and a nitrogen inlet was charged with compound (6) (16.6 g, 0.08 mol), N-hydroxysuccinimde (11.04 g, 0.096 mol), magnesium monoperoxyphthalate (80% technical grade; 110 g,-2. 3 equivalent of active oxidant) and CH2C12 (180 mL). The resulting white suspension was stirred for 5 h at 20-25°C. The reaction mixture was filtered and the filter-cake was slurried and washed with CH2C12 (3 x 300 mL). The slurry was filtered and the organic phase was washed with water (300 mL). The organic layer was then stirred twice with 20% aq. K2CO3 solution (2 x 300 mL) for 10 min to remove phthalic acid, and finally with brine (300 mL). The organic layer was dried over anhydrous Na2S04 and concentrated to give the title compound (10) as a viscous product (18.0 g, 82%) which solidified on standing. In another alternative synthesis of (10), a solution of trifluoroacetic anhydride (2 mL, 14.5 mmol) in CHUCK (10 mL) was added to a stirred suspension of urea-hydrogen peroxide complex (2.74 g, 29.1 mmol) and sodium bicarbonate (4 g, 48.5 mmol) in anhydrous CH2C12 (80 mL) under a nitrogen atmosphere at 0°C. The resulting mixture was allowed to stir at 0°C for 30 min. N-Hydroxysuccinimide (1.1 g, 9.7 mmol) was added to the reaction mixture at 0°C, followed by the addition of a solution of compound (6) (1 g, 4.85 mmol) in CH2C12 (10 mL), then the reaction mixture was warmed to ambient temperature with stirring for 16 h. The reaction mixture was decanted and the solvent was removed in vacuo. The colorless residue was dissolved in ethyl acetate (100 mL) and washed with water (1 x 50 mL) and brine (1 x 50 mL). The organic layers were pooled and dried over anhydrous magnesium sulfate and the solvent was removed in vacuo to afford the title compound (10) as a clear oil (1 g, 75%), which solidified after pumping under high vacuum. In another alternative synthesis of (10), a solution of acetic anhydride (2 mL) in CH2Cl2 (10 mL) was added to a stirred suspension of urea-hydrogen peroxide complex (2.74 g, 29.1 mmol) and sodium bicarbonate (4 g, 48.5 mmol) in anhydrous CH2CI2 (80 mL) under a nitrogen atmosphere at 0°C. The resulting mixture was allowed to stir at 0°C for 30 min. N-Hydroxysuccinimide (1. 1 g, 9.7 mmol) was added to the reaction mixture at 0°C, followed by the addition of a solution of compound (6) (1 g, 4.85 mmol) in CH2C12 (10 mL), then the reaction mixture was stirred at 40 °C for 16 h. The reaction mixture was decanted and the solvent was removed in vacuo. The colorless residue was dissolved in ethyl acetate (100 mL) and washed with water (1 x 50 mL) and brine (1 x 50 mL). The organic layers were pooled and dried over anhydrous magnesium sulfate and the solvent was removed in vacuo to afford the title compound (10) as a clear oil which solidified after pumping under high vacuum. In another alternative synthesis of (10), to a well-stirred suspension of urea- hydrogen peroxide complex (47 g, 0.5 mol) and N-hydroxysuccinimide (13. 8 g, 0.12 mol) in dichloromethane (100 mL) was added solid maleic anhydride (29.4 g, 0.3 mol). The mixture was stirred at room temperature for 15 min and then a solution of compound (6) (20.6 g, 0.1 mol) in dichloromethane (50 mL) was added slowly over a period of 15 min. The reaction proceeded with an exotherm that could be controlled by using a water bath for cooling. The reaction mixture was stirred at 20-25°C for 4 h, during which time a white precipitate formed. The reaction mixture was diluted with water (200 mL) and the phases separated. The aqueous layer was extracted with dichloromethane (200 mL), the combined organic layers were washed with brine (200 mL) and dried over anhydrous Na2S04. The solvent was removed in vacuo to afford the title compound (10) as a white crystalline solid (20.0 g, 73% yield). In still another alternative synthesis of (10), N-hydroxysuccinimide (2.3 g, 20 mmol) was added to a solution of compound (8) (1 g, 4 mmol) in CH2Cl2 (10 mL) and the reaction mixture cooled to 0°C. A solution of 32% (v/v) peracetic acid in acetic acid (0.92 g, 12 mmol) was added dropwise over a period of 10 min, then the solution was allowed to stir at room temperature for 3 h. The reaction mixture was diluted with ether (50 mL) and washed with water (2 x 10 mL), saturated sodium bicarbonate solution (10 mL) and brine (10 mL), then dried over anhydrous sodium sulfate, filtered, and concentrated in vacuo to give the title compound (10) as a as colorless oil (0.9 g, 81%). After trituration with hexane (20 mL) the product solidified to provide a white solid. In still another alternative synthesis of (10), N-hydroxysuccinimide (230 mg, 2 mmol) was added to a solution of compound (8) (248 mg, 1 mmol) in CH2C12 (10 mL) was added and the reaction mixture cooled to 0°C. m-Chloroperbenzoic acid (670 mg, 3 mmol, commercial grade: 77% in water) was added over a period of 10 min and the mixture allowed to stir at room temperature for 16 h. The reaction mixture was diluted with ether (50 mL) and washed with water (2 x 10 mL), saturated sodium bicarbonate solution (10 mL) and brine (10 mL). The organic layer was dried over anhydrous sodium sulfate, filtered and concentrated in vacuo to give the title compound (10) together with m-chlorobenzoic acid. The crude product mixture was purified by column chromatography on silica gel, eluting with 4: 6 EtOAc: hexane. Residual m-chlorobenzoic acid was removed by repeated crystallizations from a mixture of tert-butyl methyl ether and hexane which resulted in analytically pure product (30 mg, 11%). In still another alternative synthesis of (10), a 500-mL three-neck flask equipped with a mechanical stirrer, teflon-coated thermocouple and addition funnel was charged with compound (8) (20.6 g, 0.1 mol), N-hydroxysuccinimde (23.0 g, 0.2 mol) and CH2Cl2 (80 mL). The reaction mixture was cooled to 0°C and peracetic acid (32% solution in acetic acid, 16.7 g, 55 mL, 0.22 mol) added dropwise to the reaction mixture at a rate such that the temperature remained below 5°C. Upon completion of the addition, the reaction mixture was stirred for 4.5 h, maintaining the temperature at or below 15°C. The reaction mixture was then cooled to 0°C and neutralized with 10% aqueous K2C03 until the pH of the reaction mixture was-7. The mixture was then extracted with CHzClz (2x100 mL) and the combined CHUCK phases were washed with brine and dried over anhydrous sodium sulfate. The organic phase was then concentrated, affording the crude product as a white solid (20.2 g, 75%). This solid was recrystallized by dissolution in isopropanol (41 mL), warming the mixture to 40°C to afford a homogeneous solution. The solution was cooled to 0°C over two hours and the product was filtered and dried, resulting in recovery of the title compound (10) as a white solid (16 g, 79%) having a melting point of 54-56°C.
References:
WO2005/66122,2005,A2 Location in patent:Page/Page column 43-46
6066-82-6
787 suppliers
$5.00/10g
![Propanoic acid, 2-methyl-, 1-[[[(1,1-dimethylethyl)thio]carbonyl]oxy]ethyl ester](/CAS/20210305/GIF/860035-08-1.gif)
860035-08-1
0 suppliers
inquiry
860035-10-5
17 suppliers
$448.00/1g
860035-07-0
2 suppliers
inquiry
860035-10-5
17 suppliers
$448.00/1g
6066-82-6
787 suppliers
$5.00/10g
![Propanoic acid, 2-methyl-, 1-[[[4-(methylsulfonyl)phenoxy]carbonyl]oxy]ethyl ester](/CAS/20210305/GIF/1207383-11-6.gif)
1207383-11-6
0 suppliers
inquiry
860035-10-5
17 suppliers
$448.00/1g
6066-82-6
787 suppliers
$5.00/10g
860035-07-0
2 suppliers
inquiry
937-14-4
361 suppliers
$15.00/5G
860035-10-5
17 suppliers
$448.00/1g
535-80-8
418 suppliers
$5.00/5g