
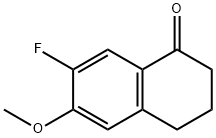
1(2H)-Naphthalenone, 5-fluoro-3,4-dihydro-6-methoxy- synthesis
- Product Name:1(2H)-Naphthalenone, 5-fluoro-3,4-dihydro-6-methoxy-
- CAS Number:88288-05-5
- Molecular formula:C11H11FO2
- Molecular Weight:194.2
Yield:125067-57-4 1.11 g ,88288-05-5 851 mg
Reaction Conditions:
Stage #1: 6-methoxy-1,2,3,4-tetrahydronaphthalenewith tetra-N-butylammonium tribromide in methanol;dichloromethane at 20; for 1.5 h;
Stage #2: with n-butyllithium in hexane at -78; for 0.25 h;Inert atmosphere;Overall yield = 3.98 mg; Further stages;
Steps:
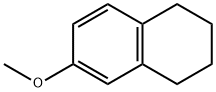
26 Mixture of 6-fluoro-7-methoxy-1,2,3,4-tetrahydronaphthalene and 5-fluoro-6-methoxy-1,2,3,4-tetrahydronaphthalene
A mixed solution of 6-methoxy-1,2,3,4-tetrahydronaphthalene (10 g) and tetrabutylammonium tribromide (29.7g) in methylene chloride (250 mL) and methanol (150 mL) was stirred for 1.5 hours at room temperature. Thereafter,the reaction solution was diluted with MTBE and was washed sequentially with water and saturated saline. The organiclayer was dried over sodium sulfate and was filtrated, and the obtained filtrate was concentrated under a reducedpressure, and thereafter, the solution was replaced with heptane to give the title compound (14.7 g). The obtainedcompound was used in the next reaction without purification.Under a nitrogen atmosphere, to a solution of the compound (6.85 g) prepared in Example 24 in THF, a solution(12.5 mL) of 2.5 M n-butyllithium in hexane was added dropwise at -78°C, and the mixture was stirred at -78°C for 15minutes. The reaction solution was treated with a solution (30 mL) of N-fluorobenzenesulfonamide (9.84 g) in THF, andthereafter, the mixture was stirred at -78°C for 30 minutes. Thereafter, the mixture was carefully treated with water (20mL), and the obtained mixture was returned to room temperature. The mixture was extracted with ethyl acetate, and theorganic layer was dried over sodium sulfate, and thereafter, the obtained solution was concentrated under a reducedpressure. The obtained residue was purified by using silica gel column chromatography (10 - 50% methylene chlo-ride/hexane) to give the title compound (3.98 mg). The obtained compound was used in the next reaction without furtherpurification. To a mixture of the compound (3.97 g) prepared in Example 25 and glacial acetic acid (36 mL), a solution ofchromium trioxide (5.06 g) in glacial acetic acid (36 mL) and water (18 mL) were added dropwise at 0°C, and the obtainedmixture was returned to room temperature for over 15 minutes. Thereafter, the reaction solution was concentrated undera reduced pressure, and the obtained mixture was poured to ethyl acetate, and the mixture was washed sequentiallywith water and saturated sodium bicarbonate aqueous solution. The organic layer was dried over sodium sulfate, andthe obtained solution was concentrated under a reduced pressure, and the obtained residue was purified by using silicagel column chromatography (0 - 20% ethyl acetate/hexane) to give 7-fluoro-6-methoxy-3,4-dihydronaphthalen-1(2H)-one(1.11 g). The remaining fraction was concentrated under a reduced pressure, and the obtained residue was purified byusing silica gel column chromatography (methylene chloride) to give 5-fluoro-6-methoxy-3,4-dihydronaphthalen-1(2H)-one (851 mg). The physical properties of each of the title compounds were as follows. 7-Fluoro-6-methoxy-3,4-dihydronaphthalen-1 (2H)-one;1H -NMR (CDCl3): δ 7.72, 6.75, 3.94, 2.91, 2.64, 2.13.5-Fluoro-6-methoxy-3,4-dihydronaphthalen-1(2H)-one;1H -NMR (CDCl3): δ 7.86, 6.92, 3.95, 2.96, 2.61, 2.13.
References:
EP3228615,2017,A1 Location in patent:Paragraph 0156; 0157; 0158
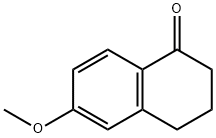
1078-19-9
379 suppliers
$6.00/5g
88288-05-5
7 suppliers
inquiry
