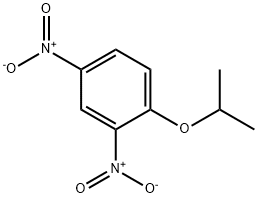
Benzene, 1-(1-methylethoxy)-2,4-dinitro- synthesis
- Product Name:Benzene, 1-(1-methylethoxy)-2,4-dinitro-
- CAS Number:10242-16-7
- Molecular formula:C9H10N2O5
- Molecular Weight:226.19
Yield:10242-16-7 90%
Reaction Conditions:
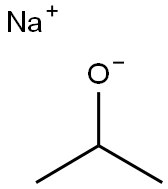
Stage #1: isopropyl alcoholwith sodium hydride
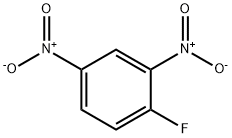
Stage #2: 1-chloro-2,4-dinitro-benzene at 80 - 172; under 760.051 - 9308.91 Torr; for 22 h;Heating / reflux;
Steps:
IV; V Example IV Preparation of 1-isopropoxy-2,4-dinitrobenzene Under Ambient Conditions
Preparation of the alkoxynitroarene 1-isopropoxy-2,4-dinitrobenzene was chosen as the branched, secondary alkoxide of the sodium isopropoxide starting material is more hindered than the NaO- utilized in the preparation of DNAN. Thus, it was hypothesized, consistent with chemical theory, that any effect of the reaction conditions on the rate of reaction would be amplified. [0057] A stock solution of sodium isopropoxide was prepared by adding 6.0 grams of sodium hydride (0.25 moles) to 400 ml of reagent grade isopropanol. The stock solution was prepared under a stream of nitrogen gas with constant agitation. The slurry became difficult to stir near the end of the hydride addition. After all of the hydride had been added, heat was applied to cause the reaction to completely proceed into solution. This solution was then set aside. [0058] One hundred mg of reagent grade isopropanol was placed in a 500 ml, three-necked round bottom flask equipped with a magnetic stir bar, argon inlet, condenser and thermocouple. To this solvent, 26.0 grams of 1-chloro-2,4-dinitrobenzene (CDB) was dissolved with gentle heating. Complete solution was observed at 80° C. To this solution, 200 ml, 0.97 equivalents of the pre-prepared sodium isopropoxide solution in isopropanol was added. The isopropoxide solution was added as rapidly as possible such that the contents of the reaction remained in the flask. Three ml samples were taken as soon as the combination had been made and subsequently at periodic intervals thereafter. The intervals were frequent at the beginning of the reaction but were less frequent at later times during the reaction. The sampling intervals are indicated on the GC/MS. A portion of each sample was subjected directly for Gas Chromatography/Mass Spectrometry (GC/MS) analysis. The samples were evaporated, dissolved in deuterated chloroform and subjected to Proton Nuclear Magnetic Resonance Spectroscopy (NMR). [0059] After refluxing a total of 22 hours, the contents of the reactor were poured onto 1 liter of ice and water. An additional 1 liter of water was then added to decrease the solubility of the product. The yellow brown solid was collected on a glass frit and washed three times with distilled water. After air drying, 15.0 grams of solid remained. Proton NMR revealed the solid to be 88% desired 1-isopropoxy-2,4-dinitrobenzene and 12% unconverted 1-chloro-2,4-dinitrobenzene. [0060] The GC/MS which resulted was compiled and plotted over time with respect to percent conversion. The data are summarized in FIG. 5. This data was verified by proton NMR as well (not shown). As is evident, the percent conversion was approximately 84% immediately upon mixing of the reagents. The additional approximately 4% conversion after about twenty-four hours may be attributed to decreased reaction time when the alkoxide (i.e., the conjugate base of sodium isopropoxide) to nitroaromatic (i.e., CDB) ratio reached approximately 1.0. A stock solution of sodium isopropoxide was prepared by adding 6.0 grams of sodium hydride (0.25 moles) to 400 ml of reagent grade isopropanol. The stock solution was prepared under a stream of nitrogen gas with constant agitation. The slurry became difficult to stir near the end of the hydride addition. After all of the hydride had been added, heat was applied to cause the reaction to completely proceed into solution. This solution was then set aside. [0062] Twenty-six grams (0.129 moles) of 1-chloro-2,4-dinitrobenzene was dissolved in 100 grams of isopropanol. This solution was subsequently placed in a 300 ml capacity Parr high pressure reactor equipped with a pressure gauge, a rupture disk, a pressure relief valve, a mechanical stirrer and a bottom sampling tube with high pressure valve. To this pale yellow homogenous solution was added, in one portion, 200 ml of the pre-prepared isopropoxide solution (3.0 grams, 0.125 mole, 0.97 mole equivalents). Three ml samples were taken as soon as the reagents had been combined and subsequently at periodic intervals thereafter as indicated on the GC/MS plot. A portion of each sample was subjected directly for Gas Chromatography/Mass Spectrometry (GC/MS) analysis. The samples were evaporated, dissolved in deuterated chloroform and subjected to Proton Nuclear Magnetic Resonance Spectroscopy (NMR). [0063] The reaction temperature was increased to 172° C. over the course of one hour. The pressure increased to 180 psi. The reaction was allowed to remain under these conditions for another half hour after which the heat was removed. The reaction was subsequently allowed to cool overnight. The contents of the reactor were subsequently poured into 1 liter of water and the solids collected on a glass frit. The solids were washed with three 100 ml portions of distilled water. After air drying, the yellowish-green solid weighed 18.1 grams. Proton NMR revealed a purity of 95%. The samples pulled during the run accounted for approximately 5 grams of starting material resulting in a yield of approximately 90%. [0064] The Gas Chromatography/Mass Spectrometry (GC/MS) analysis which resulted was compiled and plotted over time with respect to percent conversion. The data are summarized in FIG. 6. This data was verified by NMR as well (not shown). As is evident, the percent conversion was approximately 87% immediately upon mixing of the reagents. The additional approximately 8% conversion over the subsequent approximately 2.5 hours may be attributed to decreased reaction time when the alkoxide (i.e., the conjugate base of sodium isopropoxide) to nitroaromatic (i.e., CDB) ratio reached approximately 1.0. When compared to the reaction carried out under ambient pressure conditions, it is evident that the remaining conversion occurred much more quickly in the increased temperature and pressure reaction (i.e., in approximately 2.5 hours rather than twenty-four hours). [0065] A comparison of the percent conversion between examples IV and V revealed that there was an approximately 2% overall increase in the desired product yield when the reaction was carried out under increased pressure conditions. While not being held to any one theory, the inventors have, at present, attributed this increased product yield to the kinetics of the reaction including the increased rate of reaction caused by the addition of pressure when the ratio of alkoxide to nitroaromatic reached 1.0 or less.
References:
US2004/133046,2004,A1 Location in patent:Page 6-7
119-27-7
2 suppliers
$658.00/2500g
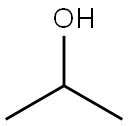
67-63-0
1550 suppliers
$17.00/25ML
10242-16-7
2 suppliers
inquiry
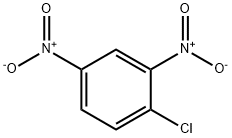
97-00-7
12 suppliers
$17.67/10gm:
10242-16-7
2 suppliers
inquiry