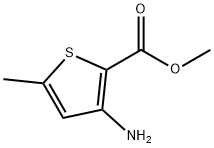
METHYL 3-AMINO-5-METHYLTHIOPHENE-2-CARBOXYLATE synthesis
- Product Name:METHYL 3-AMINO-5-METHYLTHIOPHENE-2-CARBOXYLATE
- CAS Number:76575-71-8
- Molecular formula:C7H9NO2S
- Molecular Weight:171.22

4786-20-3
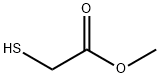
2365-48-2
76575-71-8
General procedure for the synthesis of methyl 3-amino-5-methylthiophene-2-carboxylate from 2-butenenitrile and methyl mercaptoacetate: a mixture of cis- and trans-2-butenenitrile (298.00 g, 4.44 mol) was mixed with pyridine (61.49 g, 0.78 mol) and the resulting mixture was cooled to 0 °C. Chlorine gas (314.92 g, 4.44 mol) was passed at a rate of 60-65 g/h while maintaining the internal temperature at 10-13 °C. After addition, the reaction mixture was slowly warmed to room temperature and stirred overnight. The resulting oil was dissolved in dichloromethane (1.5 L) to allow the solution to pass through a silica gel column for initial purification. The filtrate was concentrated under vacuum to give the complex reaction mixture (508.61 g) as a brown oily substance. This oily substance (493.80 g) was mixed with methyl mercaptoacetate (422.77 g, 3.98 mol), and the resulting solution was slowly (1.5 h) added dropwise to a methanol (2.2 L) slurry of potassium carbonate (1651 g, 11.95 mol) at 100 °C. The final slurry was allowed to warm to room temperature and stirred at ambient temperature for 5 hours. The solid salt was filtered out and washed with methanol (2L). Combine the organic solutions and evaporate the solvent. The residue was redissolved in ethyl acetate (3L) and subjected to secondary purification via silica gel column. The silica gel column was washed with additional ethyl acetate (2L). All organic phases were combined and the solvent was evaporated under vacuum to give a brown oil (627 g). The mixture was vacuum distilled at 6 mmHg under reduced pressure. The fraction collected at 138°C was crystallized in a receiving flask (303.07 g). The target product methyl 3-amino-5-methylthiophene-2-carboxylate (179.00 g, 26%) was obtained as white crystals by recrystallization from methanol. The structure of the product was confirmed by 1H NMR (CDCl3, 400 MHz) and 13C NMR (CDCl3, 100 MHz).
4786-20-3
80 suppliers
$9.00/1g
2365-48-2
370 suppliers
$14.00/25g
76575-71-8
109 suppliers
$35.00/1g
Yield:76575-71-8 26%
Reaction Conditions:
Stage #1: but-2-enenitrilewith pyridine;chlorine at 0 - 13;
Stage #2: Methyl thioglycolatewith potassium carbonate in methanol at 10 - 20; for 5 h;
Steps:
4
A mixture of cis- and frans-crotononitrile 4a (298.00 g, 4.44 mol) was mixed with pyridine (61.49 g, 0.78 mol) and the resulting mixture was cooled to 00C. Chlorine (314.92 g, 4.44 mol) was passed through the mixture at a rate of 60-65 g/h to maintain the internal temperature at 10-13 0C. When the addition was complete the reaction mixture was allowed to warm up to room temperature and mixed overnight. The resulting oil was dissolved in CH2CI2 (1.5 L) and the solution was passed through a silica gel plug. The filtrate was concentrated in vacuo to give a complex reaction mixture (508.61 g) as brownish oil. This oil (493.80 g) was mixed with methyl thioglycolate (422.77 g, 3.98 mol) and the resulting solution was added slowly (1.5 h) to a slurry of K2CO3 (1651 g, 11.95 mol) in methanol (2.2 L) at 100C. The final slurry was allowed to warm up to room temperature and was stirred at ambient temperature for 5 h. Salts were filtered out and washed with methanol (2 L). The organic solutions were combined and evaporated. The residue was redissolved in ethyl acetate (3 L) and passed through a silica gel plug. The plug was washed with extra ethyl acetate (2 L). The solutions were combined and the solvent was evaporated in vacuo to give brown oil (627 g). The mixture was vacuum distilled at 6 mmHg. An oil that distilled at 138 0C crystallized in the receiving flask (303.07 g). Recrystallizion from methanol yielded 4b (179.00 g, 26%) as white crystals. 1H NMR (CDCI3, 400 MHz) δ 6.25 (s, 1H), 5.30 (bs, 2H), 3.81 (s, 3H), 2.37 (s, 3H). 13C NMR (CDCI3, 100 MHz) 5 165.2, 154.5, 147.2, 119.0, 99.3, 51.4, 16.5.
References:
WO2007/23382,2007,A2 Location in patent:Page/Page column 64