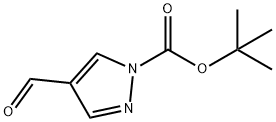
tert-Butyl 4-forMyl-1H-pyrazole-1-carboxylate synthesis
- Product Name:tert-Butyl 4-forMyl-1H-pyrazole-1-carboxylate
- CAS Number:821767-61-7
- Molecular formula:C9H12N2O3
- Molecular Weight:196.2
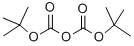
24424-99-5
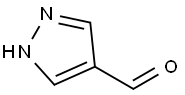
35344-95-7
821767-61-7
Step A: Di-tert-butyl dicarbonate (Boc2O, 2183 mg, 10.0 mmol) was slowly added to a stirring solution of 1H-pyrazole-4-carboxaldehyde (961 mg, 10.0 mmol) acetonitrile (30 mL) at room temperature, followed by the addition of 4-dimethylaminopyridine (DMAP, 61.1 mg, 0.500 mmol) as a catalyst. The reaction mixture was stirred at room temperature overnight. After completion of the reaction, the mixture was concentrated to dryness under reduced pressure. The residue was extracted by partitioning with ethyl acetate (30 mL) and water (30 mL). The organic phase was separated and washed sequentially with 0.5 N HCl (30 mL) and saturated saline (30 mL). The organic phase was dried over anhydrous magnesium sulfate (MgSO4), filtered and concentrated to give the crude product. The crude product was purified by chromatography on a Biotage 40M silica gel column using hexane/ethyl acetate (4:1, v/v) as eluent. Fractions containing the target product were collected, combined and concentrated under reduced pressure to afford tert-butyl 4-formyl-1H-pyrazole-1-carboxylate (1.4 g, 71% yield) as a solid.
24424-99-5
871 suppliers
$13.50/25G
35344-95-7
202 suppliers
$32.00/1g
821767-61-7
76 suppliers
$65.00/100mg
Yield:821767-61-7 98 %
Reaction Conditions:
in acetonitrile at 25;
Steps:
IV.6 Synthesis of tert-butyl 4-formyl-1H-pyrazole-1-carboxylate.
Di-tert-butyl dicarbonate (4.54 g, 20.81 mmol) was added to a solution of 1H-pyrazole-4-carbaldehyde (2.0 g, 20.81 mmol) in MeCN (21 ml) and the mixture was stirred at room temperature for 18 h. The reaction mixture was concentrated in vacuo to give an oil, which was diluted with water (20 mL) and extracted with EtOAc (3 x 20 mL). The organic phases were combined, washed with HC1 (0.5M aq., 20 mL), brine (20 mL), passed through a phase separator cartridge and concentrated in vacuo to give the title compound (4.04 g, 98 %) as a pale yellow solid. 1 H NMR (400 MHz, DMSO) δ 9.93 (s, 1H), 9.04 (s, 1H), 8.22 (s, 1H), 1.61 (s, 9H).
References:
WO2022/197763,2022,A1 Location in patent:Paragraph 0356
199003-22-0
60 suppliers
$45.00/50mg
821767-61-7
76 suppliers
$65.00/100mg
24424-99-5
871 suppliers
$13.50/25G
821767-61-7
76 suppliers
$65.00/100mg