概述[1]
达芦那韦是美国强生公司的子公司TibotecTherapeutics公司研制的蛋白酶抑制剂,2006年7月在美国首次上市,商品名为Prezista,该药常与利托那韦以及其他抗逆转录药物联合用于成人HIV感染者的后期治疗。2020年2月4日公布的最新研究表明达芦那韦达芦那在300微摩尔浓度下,能显著抑制新冠病毒复制,与未用药物处理组比较,抑制效率达280倍,具有抗新型冠状病毒活性。
作用机理[2]
达芦那韦为蛋白酶抑制药,能够阻止感染性病毒颗粒的形成,其主要经CYP3A4代谢,临床使用达芦那韦一般与利托那韦联用,可增加前者的血药浓度。HIV感染者口服达芦那韦/利托那韦600/100mg后,2~4h达到血药浓度峰值,达芦那韦血浆蛋白结合率大约为95%。
达芦那韦可选择性地抑制被感染细胞中的HIV编码Gag-Pol多聚蛋白,阻 止成熟病毒粒子的形成。体内和体外实验均已证明:达芦那韦对已产生耐药性 的HIV病毒仍具有很强的抗病毒活性,且病毒对达芦那韦产生耐药性的倾向较低。
合成方法[1]
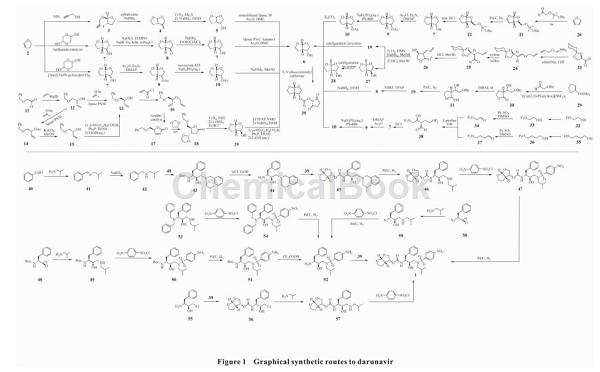
A法:首先关键中间体6与N,N'-二琥珀酰亚胺基碳酸酯反应得到39。苯甲醛(40)与异丁基胺缩合得到41,41经硼氢化钠还原得到42,42与商品化试剂(2S,3S)-1-苄基-2,3-环氧正丙基氨基甲酸叔丁酯(48)经加成反应得到43,43在盐酸作用下脱去保护基得到44,44与39经取代反应得到45,45经催化氢化脱苄基得到46,46与对硝基苯磺酰氯经取代反应得到47,47经催化氢化将硝基变为氨基制得达芦那韦(1),总收率为32%(以48为起始原料计)。
B法:48与异丁基胺反应得到49,49与对硝基苯磺酰氯经取代反应得到50,50经钯炭催化氢化得到51,51在三氟乙酸作用下脱去保护基得到52,最后52与39经取代反应制得1,总收率为80%(以48为起始原料计)。
C法:(2R,3S)-1-(异丁基氨基)-3-(二苄基氨基)-4-苯基-2-丁醇(53)与对硝基苯磺酰氯经取代反应得到54,54经催化氢化得到52,52与39经取代反应制得1,总收率为65%(以53为起始原料计)。
D法:(2R,3S)-1-氯-3-氨基-4-苯基-2-丁醇(55)与39发生亲核取代反应得到56,56与异丁基胺经取代反应得到57,57与对氨基苯磺酰氯经亲核取代反应制得1,总收率为80%(以55为起始原料计)。
E法:(2S)-[(1S)-叠氮基-2-苯乙基]环氧乙烷(58)与异丁基胺经缩合反应得到59,59经对硝基苯磺酰氯磺酰化、钯炭催化氢化得到52,52与39经亲核取代制得1,总收率为60%~66%(以58为起始原料计)。
药代动力学研究[2]
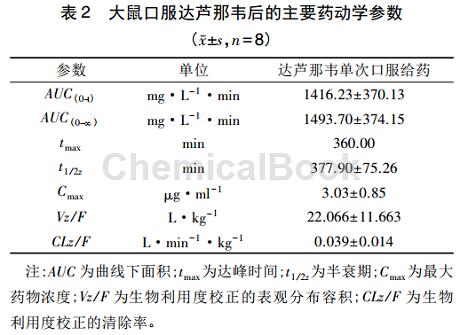
大鼠灌服达芦那韦后的药动学大鼠灌服达芦那韦后各时间点所测得的血药浓度-时间曲线见图3。血药浓度-时间数据采用DAS2.0(DrugAndStatistics)软件计算得到药动学参数。结果见表2。
制剂[3]
无定型达芦那韦100g,乳糖52g,微晶纤维素24g,低取代羟丙基甲基纤维素20g,淀粉 9g,硬脂酸镁1.5g。按下述制备方法制备1000片。
步 无定型达芦那韦过100目筛,乳糖、微晶纤维素、低取代羟丙基甲基纤维 素、硬脂酸镁过60目筛;
第二步 淀粉用纯化水制备为13%的淀粉浆;
第三步 取处方量的无定型达芦那韦,加入三分之一处方量的乳糖,混合均匀,加入三 分之一的淀粉浆,制软材;45℃烘干;
第四步 第三步所得,加入剩余处方量的乳糖,处方量的微晶纤维素、低取代羟丙基甲 基纤维素 ,混匀,加入余量的淀粉浆,制软材,整粒,45℃烘干;
第五步 第四步所得,加入处方量的硬脂酸镁,压片。
主要参考资料
[1]刘鸿,展鹏,刘新泳.达芦那韦合成路线图解[J].中国药物化学杂志,2013,23(01):72-74.
[2]张小瑞,王力彬,李鑫.LC-MS/MS测定大鼠血浆中的达芦那韦及其药动学研究[J].中国药师,2020,23(01):91-93+171.
[3] [中国发明,中国发明授权] CN201210205848.9 制备达芦那韦无定形物的方法