背景及概述[1]
柔红酮是表柔比星中间体。表柔比星是由辉瑞开发的用于治疗乳腺癌、肺癌、肝癌的蒽环类抗肿瘤抗生素,1984年在欧洲上市,1999年在美国上市。在治疗白血病、淋巴瘤和各种实体瘤(包括乳腺癌、非小细胞肿瘤、子宫颈癌和头颈癌)中有着广泛的应用。其作用机制是直接嵌入DNA核碱对之间,干扰转录过程,阻止mRNA的形成,从而抑制DNA和RNA的合成。此外,表阿霉素对拓扑异构酶Ⅱ也有抑制作用。为一细胞周期非特异性药物,对多种移植性肿瘤均有效。与阿霉素相比,疗效相等或略高,但对心脏的毒性较小。
制备[1]
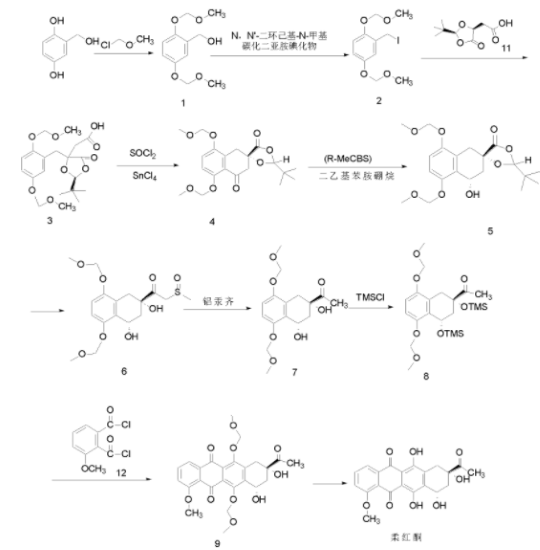
中间体1的制备
将2,5-二羟基苄醇(14.0g,0.1mol)溶于1.4L二氯甲烷中,氮气保护,加入二异丙基乙胺(62.8mL,0.38mol)和氯甲基甲醚(21.3mL,0.28mol),保持搅拌回流22h;反应完毕,降温,将反应液倒入1.4L质量分数为5%的碳酸氢钠溶液中,搅拌约10min,分液,水相用二氯甲烷(3×500ml)萃取,收集合并有机相,依次用1.4L水洗涤,1.4L饱和食盐水洗涤有机相,有机相再用无水硫酸钠干燥,过滤,滤液经减压浓缩得到棕色油状物,加入70ml石油醚打浆约30min,抽滤,减压干燥得白色固体即为中间体1,摩尔收率97.8%,HPLC纯度98.2%。
中间体2的制备
将中间体1(18.0g,78.9mmol,HPLC纯度98.2%)置于500mL反应瓶中,加入溶有N,N'-二环己基-N-甲基碳化二亚胺碘化物(54.7g,158mol)的四氢呋喃溶液270mL,30℃下,氮气保护,避光搅拌3h,减压蒸除溶剂,残余物溶于在200mL正己烷中,用水(100mL×3)洗涤三次,水相用150ml正己烷萃取,收集有机相并用无水硫酸钠干燥,抽滤,滤液减压浓缩,用二氯甲烷和正己烷混合溶剂重结晶得中间体2,摩尔收率96.0%,HPLC纯度98.4%。
中间体3的制备
将酸11(5.0g,24.7mmol)溶于200mL正己烷中,在-78℃,氮气保护下缓慢地滴入130mL溶有双三甲基硅基胺基锂(8.27g,49.4mmol,)的四氢呋喃溶液,20min后,加入50mL溶有中间体2(12.5g,37.0mmol,HPLC纯度98.4%)的正己烷溶液。-78~-50℃下搅拌1h,升温至-30℃搅拌20h,反应完毕,将反应液倒入300mL 1mol/L盐酸中,用150mL×3乙醚萃取三次,收集有机相,有机相用MgSO4干燥,减压蒸除溶剂,将残余物溶于250mL二氯甲烷,用125mL×3饱和碳酸氢钠溶液洗涤三次,水相用150mL×3乙醚萃取三次,收集合并有机相,MgSO4干燥,减压浓缩得类白色固体即为中间体3,摩尔收率89.2%,HPLC纯度96.4%。
中间体4的制备
将中间体3(15.0g,36.4mmol,HPLC纯度97.2%)的置于反应瓶中,在室温下加入225mL二氯甲烷,氮气保护下加入氯化亚砜(11.2mL,0.164mol),加热回流反应12h,冷却至室温后加入四氯化锡(27.0mL,0.146mol),在20℃下再搅拌1h,反应完毕,降温至0℃,加入225g碎冰,分液收集水相,水相在0℃下用二氯甲烷120mL×3萃取三次,收集合并有机相,分别用400mL饱和NaHCO3溶液和400mL饱和氯化钠溶液洗涤,有机相用无水MgSO4干燥,减压浓缩,得中间体4粗品,摩尔收率96.4%,HPLC纯度87.1%;中间体4粗品再用四氢呋喃和正己烷体积比为1:3的混合溶液重结晶,得中间体4,摩尔收率88.3%,HPLC纯度99.3%。mp:158℃,[α]20D=+48.4(c=0.48,CHCl3)]。
中间体5的制备
氮气保护下向反应瓶中加入80mL四氢呋喃,开搅拌,加入N,N-二乙基苯胺硼烷(5.0mL,27.9mmol),将催化剂(R)-2-甲基-CBS-恶唑硼烷((R)-MeCBS)(0.22g,2%)溶于80ml四氢呋喃,并流加入反应瓶中,反应液控温15~25℃,用恒压滴液漏斗滴加220mL溶有中间体4的(11.0g,27.9mmol,HPLC纯度99.3%)的四氢呋喃溶液,确保2~3h滴完,保温反应10-20min;反应完毕,控制温度低于25℃,缓慢滴加32mL甲醇,搅拌15min,减压浓缩,加入300mL二氯甲烷,控温15~25℃滴加160mL 2mol/L的硫酸,有泡沫出现,搅拌15min,加160mL水,分液,有机相依次用250mL水洗涤,250mL饱和氯化钠溶液洗涤,无水硫酸钠干燥,抽滤,滤液减压浓缩至干,再用四氢呋喃和乙醚体积比为2:3混合溶液重结晶,得中间体5,摩尔收率94.2%,HPLC纯度98.8%。
中间体6的制备
将中间体5(9.51g,24mmol,HPLC纯度98.8%)溶于500mL四氢呋喃,氮气保护下,降温至0℃,将0.1mol二甲亚砜钠盐溶于50mL二甲亚砜和四氢呋喃的混合溶液并滴入反应瓶中,在室温下搅拌30min反应完毕,加入1.25L二氯甲烷和2.0L饱和氯化铵溶液搅拌5min,静置分液,水相用1.25L×2二氯甲烷洗涤两次,合并有机相,用1.0L水洗涤有机相,用无水硫酸钠干燥有机相,过滤,滤液经减压浓缩,得中间体6,摩尔收率98.0%,HPLC纯度96.7%。
中间体7的制备
将中间体6(9.13g,23.5mmol,HPLC纯度96.7%)溶于250mL四氢呋喃和25mL水的混合溶液中,氮气保护,在室温下加入铝汞齐(10.0g,0.36mol),搅拌90min,过滤滤除固体,滤饼用少量四氢呋喃洗涤,滤液减压浓缩,向浓缩液中加入200mL乙醚和50mL水的混合溶液,搅拌,静置,分出水相,有机相用无水硫酸钠干燥,减压蒸除溶剂,浓缩液再经二氯甲烷和乙醚体积比为1:1.8的混合溶液重结晶得中间体7,摩尔收率86.4%,HPLC纯度99.0%。[α]20D=+8.0(c=1.78,氯仿)
中间体8的制备
向三口烧瓶中加入中间体7(7.0g,21.5mmol,HPLC纯度99.6%),用42mL二氯甲烷溶解,0℃,氮气保护下,滴加三乙胺(6.0mL,43mmol),然后再滴加三甲基氯硅烷(6.00mL,47.3mmol),0℃下搅拌反应1~2h,反应结束,向反应体系中加入100mL二氯甲烷稀释,反应液倒入100mL冰水中,萃取、收集有机相,依次用100mL×2水洗涤两次,100mL×2饱和食盐水洗涤两次,有机相用无水硫酸钠干燥,抽滤、滤液减压浓缩至干得中间体8,摩尔收率为98.7%,HPLC纯度98.4%。
中间体9的制备
向反应瓶中加入中间体8(10.1g,21.5mmol,HPLC纯度98.4%)的二氯甲烷溶液,分批加入三氯化铝(8.6g,64.5mmol),降温至0℃,缓慢滴入30mL溶有化合物12(5.0g,21.5mmol)的二氯甲烷溶液,0℃搅拌30min,自然升至室温,搅拌6-8h,将反应液倒入的70mL质量分数为3%HCl溶液中,0℃保温搅拌10min,室温搅拌0.5~1h,静置分液,分出有机相,水相用40mL二氯甲烷萃取,收集有机相,有机相用100mL饱和碳酸氢钠溶液洗涤,无水硫酸钠干燥,减压浓缩,浓缩液经二氯甲烷和异丙醚体积比为1:4的混合溶液重结晶,再用乙醇和乙醚体积比为1:5的混合溶液重结晶,得中间体9。摩尔收率93.9%,HPLC纯度99.7%。
柔红酮的制备
将中间体9(9.3g,19.1mmol,HPLC纯度99.7%)溶于120mL二氯甲烷,氮气保护下,降温至2-8℃,滴入20mL溶有二甲基溴化硼(4.6g,38.2mmol,)的二氯甲烷溶液,保温2-8℃搅拌2h,将反应液倒入80mL饱和碳酸氢钠溶液和120ml四氢呋喃的混合溶液,搅拌15min,分液,水相用80mL×3二氯甲烷萃取三次,收集合并有机相,有机相用160mL饱和食盐水洗涤,无水硫酸钠干燥,减压浓缩得柔红酮,摩尔收率98.2%,HPLC纯度99.5%。
参考文献
[1] [中国发明] CN201810259489.2 一种表柔比星中间体柔红酮的合成方法