卡培他滨简介
卡培他滨[1~4] (capecitabine,1),化学名为 5'-脱氧-5-氟-N-[(戊氧基) 羰基]胞嘧啶核苷,是由罗氏公司研制的口服核苷类抗肿瘤药物。本品是5-氟尿嘧啶(5-FU)的前体药物,于 1998 年 8 月首次在瑞士上市,1998 年 9 月获美国 FDA 批准用于治疗对紫杉醇和多柔比星等药物无效的晚期原发性或转移性乳腺癌;2001 年 FDA 批准本品用于治疗转移性结肠直肠癌[5] 。
制备
文献对化合物 1 的合成方法进行了归纳总结[6] 。其中,合成路线较短的为:以 D-核糖(2)为起始原料,经 1-位羟基甲醚化及 2,3-位羟基保护得 2,3-O-异亚丙基-D-呋喃型甲基糖苷(3),在吡啶作用下与对甲苯磺酰氯反应得 2,3-O-异亚丙基-5-O-对甲苯磺酰基-D-呋喃型甲基糖苷(4),经碘化钠碘代得 2,3-O-异亚丙基-5-碘-D-呋喃型甲基糖苷(4'),经 Pd/C 催化氢化还原得 2,3-O-异亚丙基-5-脱氧-D-呋喃型甲基糖苷(5),经硫酸脱保护基、乙酸酐乙酰化得 1,2,3-三-O-乙酰基-5-脱氧-D-呋喃核糖(6),再与 5-氟胞嘧啶在无水四氯化锡作用下缩合得 2',3'-二-O-乙酰-5'-脱氧-5-氟胞苷(7)。7 与氯甲酸正戊酯反应得 2',3'-二-O-乙酰-5'-脱氧-5-氟-N 4 -[(戊氧基)羰基]胞嘧啶核苷(8),再经氢氧化钠水解得终产物[7~9] 。在该路线基础上,文献报道化合物 4 可在 DMSO 中经硼氢化钠还原直接得 5 [10] ;文献 [11] 报道可用对硝基苯甲酸戊酯替代氯甲酸正戊酯;还有文献报道化合物 4 可用亚磷酸还原得 5 [12] 。上述路线总收率较低。综合分析几条合成路线,本研究参照文献[12] 合成路线进行改进与优化,设计出以下改进的合成路线(图 1)。
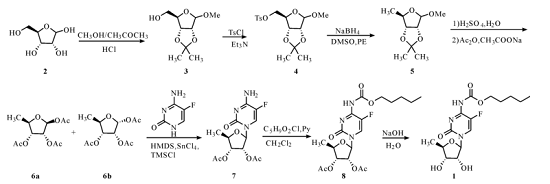
2,3-O-异亚丙基-D-呋喃甲基糖苷(3)的合成 将化合物 2(20. 0 g,0. 13 mol)、甲醇(80 ml,1. 73 mol)、丙酮(80 ml,0. 95 mol)和浓盐酸(2 ml,0. 02 mol)依次加至反应瓶中,加热至回流反应 6h,TLC[展开剂:石油醚 ∶ 乙酸乙酯(3 ∶ 1)]显示反应完全。冷却至室温,缓慢加入碳酸氢钠固体调至 pH 7~8 后,于 40 ℃减压回收丙酮和甲醇至干,得浅红棕色油状物;冷却至室温,加入二氯甲烷(DCM,40 mL)和水(40 mL),搅拌 5 min,分液,收集有机相,水相用 DCM(30 ml)萃取,合并 DCM相,用无水硫酸钠干燥,过滤,滤液减压旋干,得浅黄色油状物 3(22 g,82. 52%),不用纯化直接用于下一步反应。
2,3-O-异亚丙基-5-O-对甲苯磺酰基-D-呋喃甲基糖苷(4)的合成 将化合物 3(20. 2 g,0. 10mol)、DCM(50 mL)和三乙胺(20 mL,0. 14 mol)加至反应瓶中,冰水浴搅拌条件下控制滴加对甲苯磺酰氯(15. 6 mL,0. 12 mol) 速度控制内温 20±5℃,加完保温 20±5℃ 搅拌反应 14 h,TLC(展开剂:同 3)显示反应完全。加入水(50 mL),继续搅拌 1 h。分液,有机相用水(50 mL)洗,用无水硫酸钠干燥,过滤,滤液减压旋干,得棕红色油状物,室温放置很快成浅红棕色颜色固体 4 (34. 0 g,97. 86%)不用纯化直接用于下一步反应。
2,3-O-异亚丙基-5-脱氧-D-呋喃甲基糖苷(5)的合成 将化合物 4(33. 5 g,0. 09 mol)、DMSO(17 mL)、石油醚(50 mL)和硼氢化钠(6. 6 g,0. 17 mol)依次加至反应瓶中,室温搅拌反应 10min,然后再加热至回流搅拌反应 10 h,TLC(展开剂:同 3)显示反应完全。冷却至室温,抽滤反应液产生的白色固体,滤饼用石油醚(10 mL)淋洗;合并滤液,用水(40 mL×3)洗,用无水硫酸钠干燥,过滤,滤 液 减 压 旋 干 得 无 色 油 状 物 (17. 3 g,94. 53%)。不经纯化直接用于下步反应。
1,2,3-三-O-乙酰基-5-脱氧-D-呋喃核糖(6a&6b)的合成 将化合物 5(12 g,0. 06 mol)和H 2 SO 4 水溶液(50 mL,0. 04 mol·L-1)依次加至反应瓶中,加热至 100 ℃搅拌反应 2 h,TLC(展开剂:同 3)显示反应完全。冷却至室温,加入吡啶(约0. 25 mL)调至近中性,减压旋干得淡黄色油状物(8. 5 g,99. 40%)。
向上述淡黄色油状物中加入乙酸酐(8. 5 ml,0. 09 mol),搅拌条件下缓慢加入无水乙酸钠(4. 1g,0. 05 mol),加完室温搅拌反应 4 h;TLC(展开剂:同 3)显示反应完全;向反应液中加入冰水(30mL),用碳酸氢钠固体(约 15 g)调至近中性;加入乙酸乙酯(50 mL),搅拌 10 min,分液;乙酸乙酯相用无水硫酸钠干燥,过滤,滤液减压旋干得浅黄色油状物(14. 2 g,86. 48%)。
取适量上述油状物经硅胶柱色谱[洗脱剂:石油醚 ∶ 乙酸乙酯(3 ∶ 1)]分离纯化得单一的无色油状物 。
2',3'-二-O-乙酰-5'-脱氧-5-氟胞苷(7)的合成 将甲苯(10 mL)、5-氟胞嘧啶(1. 76 g,0. 01mol)、六甲基二硅氮烷(HMDS,2. 75 g,0. 02 mol)和三甲基氯硅烷(TMSCl,0. 15 mL,0. 1 mmol)依次加至反应瓶中,加热至回流反应 3 h。冷却至 70℃,减压蒸除有机溶剂;降至 0±5 ℃,加入化合物6a&6b(4 g,0. 02 mol)的 DCM(22 mL)溶液后,再保温缓慢滴加无水 SnCl 4 (2 ml,0. 02 mol),加毕控温 0±5 ℃搅拌反应 3 h。TLC(展开剂:同 3)显示反应完全。将反应液倒至加有碳酸氢钠固体(6. 7g,0. 08 mol)的烧杯中,搅拌下滴加 H 2 O(3. 8 ml),滴完搅拌 1. 5 h。抽滤反应过程中产生的白色固体,滤饼用 DCM(10 mL×2)淋洗,合并滤液,依次用碳酸氢钠溶液(10 mL,0. 05 mol·L-1)和 H 2 O(5mL)洗涤,用无水硫酸镁干燥,抽滤;滤液减压旋干后加异丙醇(10 mL),加热使其溶解,冷却至室温后转移至 2 ℃冰箱析晶 6 h。抽滤,滤饼用 2 ℃异丙醇(2 mL)淋洗,抽干,滤饼于 45 ℃减压旋蒸2 h 得白色粉状固体产物 7(4. 1 g,81. 2%)。
2',3'-二-O-乙酰-5'-脱氧-5-氟-N 4 -[(戊氧基)羰基]胞嘧啶核苷(8)的合成 将吡啶(1. 0mL,0. 01 mo1) 缓慢加至化合物 7(3. 7 g,0. 01mo1)的无水 DCM(18. 5 mL)溶液中,加毕,保温-5±5 ℃;滴加氯甲酸正戊酯(2. 2 mL,0. 02 mo1)的DCM(11 mL)溶液,约 1 h 加完;加毕室温搅拌反应 2 h。TLC(展开剂:同 3)显示反应完全。反应液用饱和氯化钠溶液(10 mL×2)洗涤,分出有机相,用无水硫酸钠干燥,过滤;滤液减压旋干得淡黄色油状物(4. 8 g,96. 40%)。
卡培他滨(1)的合成 将油状物 8 溶解于甲醇(14 mL)溶液中,冷却至-5±5 ℃,缓慢滴加氢氧化钠溶液(39 mL,0. 5 mol·L-1),加完继续搅拌反应 30 min。TLC(展开剂:同 3)显示反应完全用1 mol·L-1盐酸(20 mL)调至 pH 5~6,用 DCM(50mL)萃取,有机相用无水硫酸钠干燥,过滤;滤液加入活性炭 0. 1g 室温搅拌 30min;过滤;减压回收DCM 至干,得淡黄色油状物(3. 8 g);加入正己烷(90 mL)加热至溶解,于 2 ℃放置析晶 8 h。抽滤,滤饼用 2 ℃的正己烷(10 mL)淋洗,于 45 ℃减压旋蒸 2h 得白色粉状固体(3. 2 g,82. 10%),纯度99. 6%。
总结
通过本研究的合成方法合成卡培他滨 1,为工业化生产目标化合物提供了一条可行的放大生产路线,其合成路线操作简便,安全可控,成本低,总收率提升。
参考文献
[1]赵明礼,赵玉涛,张召,等.抗肿瘤药物卡培他滨的合成新方法[J].高等学校化学学报,2012,33:1 733-1 737.
[2]Yan Lou,Qian Wang,Jinqi Zheng,et al.Possible pathways of capecitabine-induced hand-foot syndrome[J].Chem Res Toxicol,2016,29(10):1 591-1 601.
[3]Justin B Renaud,Lyne Sabourin,Edward Topp,et al.Spectral counting approach to measure selectivity of high-reso-lution LC-MS methods for environmental analysis[J].Anal Chem,2017,89(5):2 747-2 754.
[4]姚刚,余艳枝.卡培他滨的合成工艺研究[J].山西医科大学学报,2012,43(1):25-29.
[5]马新成,魏长凤,岳建华.卡培他滨[J].齐鲁药事,2005,24(10):638-638.
[6]何学军,肖亚东,王德才.卡培他滨合成路线图解[J].中国医药工业杂志,2009,40(7):549-551.
[7]Fei X,Wang JQ,Miller KD,et al.Synthesis of[18 F]Xeloda as a novel potential PET radiotracer for imaging enzymesincancers[J].Nucl Med Biol,2004,31(8):1 033-1 041.
[8]Shimma N,Umeda I,Arasaki M,et al.The design and syn-thesis of a new tumor-selective fluoropyrimidine carba-mate,capecitabine[J].Bioorg Med Chem,2000,8(7):1697-1 706.
[9]Li JL,He BF,Shao LY,et al.Preparation and application of a N4-oxycarbonylcytosine derivatives[P]. WO: 2005080351,2005-09-01.
[10]Nagaraju Mekala,Murthy V R K Moturu,Rao V L N Dammalapati,et al.Safe and alternate process for the re-ductions of methanesulfonates: application in the synthesis of 1,2,3-triacetyl-5-deoxy-d-ribofuranoside[J].Org Process Res Dev,2016,20(3):609-614.
[11]陈莉莉,岑均达.卡培他滨的新合成路线[J].中国药物化学杂志,2010,20(4):275-277.
[12]李志裕,车文军,尤启冬.卡培他滨的合成[J].中国医药工业杂志,2008,39(11):804-807.