背景及概述【1】
乳腺癌是在乳腺上皮组织出现的恶性肿瘤,99%发生在女性,其发病率位居全球女性癌症之首。乳腺不是维持人体生命活动的重要器官,原位乳腺癌并不致命,但由于乳腺癌细胞丧失了正常细胞的特性,细胞之间连接松散,容易脱落。癌细胞一旦脱落,游离的癌细胞随血液或淋巴液播散至全身,形成转移性癌变,危及生命。
目前乳腺癌已成为威胁女性身心健康的常见肿瘤和“杀手”,已是当前社会的重大公共卫生问题。Ribociclib 暂译名为瑞博西尼,其他译名为瑞博西林、瑞布西利、瑞布昔利布等,代号 LEE-011。瑞博西尼的活性组分是游离碱,稳定成分是琥珀酸盐,英文化学名为 Butanedioic acid,7-cyclopentyl-N,N- dimethyl-2-{[5-(piperazin-1-yl) pyridin-2-yl]amino}-7H-pyrrolo[2,3-d]pyrimidine-6-carboxamide,中文化学名为琥珀酸 7-环戊基-N,N-二甲基-2-{[5-(哌嗪-1-基) -吡啶-2-基]氨基}-7H-吡咯并[2,3-d]嘧啶-6-羧酰胺。
琥珀酸瑞博西尼 (ribociclib succinate;)由瑞士诺华制药有限公司(Novartis Pharmaceuticals Ltd)研制,是一种细胞周期蛋白依赖性激酶 4/6(cyclin-dependent kinases4/6,CDK4/6)抑制药,与细胞周期蛋白(cyclin)结合,促进细胞周期时相转变,启动 DNA 合成,调控细胞转录。CDK4/6 抑制药可将肿瘤细胞阻滞于细胞周期的阶段(G1 期),从而起到抑制肿瘤增殖的作用。
该药适用于治疗绝经后妇女的激素受体阳性(hormone receptor,HR + ) 和人表皮细胞生长因子受体-2 阴性(human epidermal growth factor receptor-2 - ,HER2 - )的晚期或转移性乳腺癌。2016 年 8 月 9 日获美国食品药品管理局(FDA)突破性疗法资格,FDA 于 2016 年11 月 9 日接受诺华制药有限公司瑞博西尼的新药上市申请,作为一线治疗药物,与来曲唑组合用药的治疗组与安慰药联用的对照组比较,可减少疾病进展或死亡风险 (hazard ratio,HR) 达 44%[95% CI: (0429,0720);P<0.01],并显著延长所有患者群体 9.3 个月无进展生存期(progress free survival,PFS)。FDA 给予优先审查,于 2017 年 3 月 17 日正式批准上市,商品名为Kisqali。
非临床药理毒理学【1】
1.致畸、致突变 尚未对瑞博西尼进行癌病变研究。瑞博西尼无基因毒性,体外细菌回复突变 Ames试验、人淋巴细胞染色体畸变试验及体内大鼠骨髓微核致染色体断裂实验均为阴性。
2 . 对生殖能力的影响 尚未在动物中对瑞博西尼进行影响生育力的研究。在大鼠和犬重复给予喂饲瑞博西尼的毒性研究中,每天给药 1 次,连续 3 周,停药1 周,大鼠给药剂量为瑞博西尼≥75 mg·kg-1 ,犬的剂量为≥1 mg·kg-1,分别喂饲 26 及 39 周,观察到雄性动物睾丸有萎缩性变化。大鼠和犬的睾丸出现生精小管上皮细胞的退行性变性,精子减少症,附睾中管腔细胞碎片和大鼠附睾上皮细胞的空泡形成。
大鼠和犬的给药剂量其全身药物接触量,根据药物浓度-时间曲线下面积(AUC)计算,分别相当于人用最高推荐日剂量600 mg AUC 接触量的 1.4 和 0.03 倍。上述不良效应可能直接与睾丸生殖细胞抗增殖作用有关联,导致生精小管的萎缩,但在大鼠和犬停止给药 4 周后显示可逆性趋势。
3. 动物药理毒理学 在体内对犬进行心脏安全性研究,犬接触类似于患者所接受瑞博西尼 600 mg 的推荐剂量,显示与剂量和浓度相关的 QTc 间期延长。当药物接触量升高时,约为预期临床药物峰值浓度(C max )的 5 倍时,可能诱发室性期前收缩(prematureventricular contractions,PVCs)的发生。
临床药理毒理学【1】
1 .作用机制
瑞博西尼是一种细胞周期蛋白依赖性激酶 4/6(cyclin-dependent kinases 4/6,CDK4/6)抑制药,这些激酶与细胞 D-型周期蛋白(D-cyclin)结合,形成活性复合物后,对调节细胞周期进程和细胞增殖的信号通路中起关键作用。D-型周期蛋白复合物使视网膜母细胞瘤蛋白(retinoblastoma protein,pRb) 磷酸化,调节细胞周期由 G1 期向 S1 期转变,调控细胞转录,启动 DNA 合成。
而瑞博西尼将细胞周期阻滞于G1 期,从而起到抑制肿瘤增殖的作用。在体外,瑞博西尼在乳癌细胞系中,能降低 pRb 磷酸化使细胞周期停滞在 G1 期,并减缓乳癌细胞系的细胞增殖,在体内,大鼠人肿瘤细胞异种移植模型,单次给予瑞博西尼,能使肿瘤体积减少,此效应与抑制 pRb 磷酸化有相关性。在患者雌激素受体阳性乳腺癌移植瘤模型的研究中,瑞博西尼与抗雌激素药来曲唑联用,与各药单用比较,对肿瘤生长的抑制作用增加。
2. 药效学的心脏电生理学
评价瑞博西尼对晚期乳腺癌患者 QTc 间期的影响,在单次给药后和稳态时一式 3 份同步检测心电图中进行。一项纳入 267 例患者的药动学/药效学分析研究,服用瑞博西尼剂量范围为 50~1 200 mg,包括193 例患者服用 600 mg 剂量,分析结果提示,瑞博西尼对 QTc 间期的延长,与药物浓度的增加有相关性。给予 600 mg 推荐剂量,在 C max 稳态时,QTcF 间期估算值从基线的均数变化为 22.9 ms[90%CI:(21.6,24.1) ms]。
药动学
瑞博西尼在 50~1 200 mg 剂量范围内,无论是单次给药还是多次重复给药,其 C max 和 AUC 都显示超正相关增加。重复给予 600 mg,每天 1 次,一般在 8 d 后达到稳态时,瑞博西尼几何均数积蓄比为2.51(范围 0.97~6.40) [1-3] 。2.3.1 吸收 瑞博西尼给药后,达到 C max 的时间 t max 为1.0~ 4.0 h。
与空腹比较,进食高脂高热餐(3.34 ~4.18 kJ),其中,约 50%来自脂肪,35%来自碳水化合物,15%来自蛋白质,不影响对瑞博西尼的的吸收速率和程度。C max 几何均值比值(geometric mean ratio,GMR)为1.00,90% CI:(0. 898,1.11)%; AUC inf GMR 为 1. 06,90%CI:(1.01,1.12)% 。
分布
瑞博西尼在体外与人血浆蛋白的结合率约 70%,在(10~10 000) ng·mL-1范围内与药物浓度无关。在体内,瑞博西尼均等分布于红细胞和血浆内,血与血浆的比值为 1.04。群体药动学分析结果表明,稳态时表观分布容积(Vss/F)为 1 090 L。
代谢
体内外研究表明,瑞博西尼在人体内主要通过肝脏细胞色素 CYP3A4 酶进行广泛代谢。单次口服放射性标记瑞博西尼 600 mg,主要代谢途径涉及氧化[脱烷基化,C-键和(或)N-键的氧合作用及2H-键的氧化反应]和其后的结合反应。
瑞博西尼的初级代谢物通过 N-乙酰化、硫酸化、半胱氨酸共轭结合,糖基化和葡萄糖醛酸化作用产生次级结合物。在循环中主要代谢衍生物约占 44%,包括代谢物 M13(CCI284,N-羟基化物),M4(LEQ803,N-去甲基化物)和 M1(次级葡萄糖醛酸苷)等,其估算值约占总放射性的 9%,9%和 8%,分别为瑞博西尼接触量的 22%,20%和 18%。
瑞博西尼的临床疗效和安全性主要来自母体化合药,循环中代谢物的贡献率可忽略不计。从粪便和尿中回收的代谢物中,未变化的原型药物分别占 17% 和12%,代谢物 LEQ803 分别约占给药量的 14%和 4%,其他代谢物的检测量均小于 3%给药剂量。
消除
晚期癌症患者口服瑞博西尼 600 mg,在稳态时,根据蓄积比值计算,血浆有效半衰期的几何均值为 32.0 h 变异系数(coefficient of variation,CV)为63%,表观口服清除率(CL/F)几何均值为 25.5 L·h-1(CV=66%);表观血浆末端半衰期(t 1/2 )几何均值为(29. 7 ~ 54. 7) h。
健康受试者同样口服瑞博西尼600 mg,CL/F 几何均值为(39.9~77.5) L·h-1 。瑞博西尼主要地通过粪便消除,经肾脏消除的贡献率较小。6 例健康男性受试者,单次口服放射性标记瑞博西尼后,92%总放射性剂量在服药后 22 d 内被回收;粪便是排泄的主要途径,约占 69%,尿中回收 23% 。
特殊人群的药动学
肝损伤患者:根据一项群体药动学分析,其中,包括160 例肝功能正常患者和 47 例轻度肝损伤患者。轻度肝损伤患者(Child-Pugh 为 A 级),对瑞博西尼的接触量无影响。B 级肝损伤患者对瑞博西尼接触量的几何均数增加值<50%,C max 增加值为 1.50,AUC inf 为1.32;C 级严重损伤患者几何均数增加值 C max 为1.34,AUC inf 为1.29。
肾损伤患者:尚不清楚严重肾损伤患者[eGFR<30 mL· min-1·(1.73 m 2 )-1]对瑞博西尼药动学的影响一项群体药动学分析表明,轻度肾损伤患者[eGFR 为(60~<90) mL· min-1·(1.73 m 2 )-1]和中度肾损伤患者[eGFR 为(30~<60) mL· min-1·(1.73 m 2 )-1]不影响对瑞博西尼的接触量。
适应证
瑞博西尼适用于与芳香化酶抑制药联用,作为初始内分泌治疗,用于妇女绝经后激素受体 HR + 阳性,HER2 的晚期或转移性乳癌治疗。
剂量与服法
剂型与规格 口服薄膜包衣片,每片含有效成分瑞 博 西 尼 200 mg,相 当 于 琥 珀 酸 瑞 博 西 尼254.40 mg。
推荐剂量为口服 600 mg(3 片200 mg 膜包衣片),每天 1 次,连续服 21 d,接着停服7 d,28 d 为 1 个疗程。是否与食物同服均可。薄膜包衣片应整片吞服,吞咽前包衣片不可咀嚼、压碎或劈开。患者若服药后发生呕吐或缺少一次剂量,当天不可追加服用附加剂量,可按治疗时间表,在下次服药时间,继续服下一次剂量。若与来曲唑或其他芳香化酶抑制药同服,尽量在每天大约相同时间服药,在早晨。
发生不良反应需要调整服药剂量 起始服药剂量为每天 600 mg(3 片 200 mg 包衣片),首次减小剂量为每天 400 mg(2 片 200 mg 包衣片),第 2 次减小剂量为每天 200mg(1 片 200 mg 包衣片)。若需要再次减小剂量,每天小于 200 mg,应停止治疗。
不良反应 【1】
研发公司对瑞博西尼所开展的15 项 临 床 试 验 研 究,已 完 成 11 项,仅 对 代 号 为MONALEESA-2 的Ⅲ期临床研究,提供详尽的安全性数据。此项临床研究纳入 668 例绝经后晚期乳腺癌妇女患者,接受瑞博西尼联用来曲唑(简称治疗组)与安慰药加服来曲唑(简称对照组)进行对比试验,联用药组接触药物的中位时间为 13 个月,其中,有 58%患者接触时间≥12 个月。
联用组有 45%患者因不良反应需要减低服药剂量,而对照组仅为 3%。两组分别有7%和 2%因不良反应永久终止治疗,其主要原因是ALT 升高(4%)、AST 升高(3%)和呕吐(2%);此外,两组分别分别有 3 例(0.9%)和 1 例(0.3%)在治疗期间死亡,治疗组 3 例死亡的病因分别是疾病进展、3 级低钾血症和 2 级 QT 间期延长。最常见的、发生率≥20%的不良反应分别为中性粒细胞减少、恶心、疲乏、腹泻、白细胞减少、脱发、呕吐、便秘、头痛和背痛等;实验室检测>2%异常为 3 或 4 级中性粒细胞减少、白细胞减少、异常肝功能测试、淋巴细胞减少等。
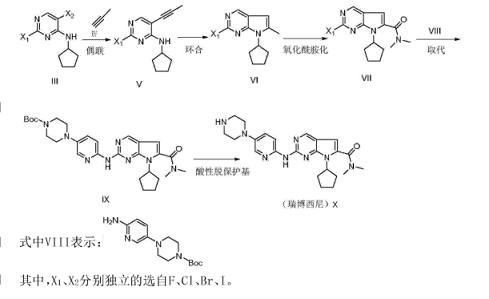
合成路线
参考文献
[1]陈本川.治疗晚期乳腺癌新药——瑞博西尼(ribociclib)[J].医药导报,2017,36(08):945-951.