背景技术
氟喹酮1983年在日本首次上市,由日本田边株式会社开发,经过二十多年的临床应用,证明是一个对面、颈部肌肉痉挛有良好疗效的松弛剂。作为肌肉松弛药,氟喹酮主要作用于脊上位中枢较广泛的部位而使肌肉紧张性亢进状态缓解。它的中枢肌肉松弛活性大约是甲苯丙醇的22-29倍,是氯美扎酮的8-10倍。与已知的肌松药相比,氟喹酮的副作用和毒性都较小,具有很好的安全性。
在本发明给出之前,氟喹酮的主要合成方法:
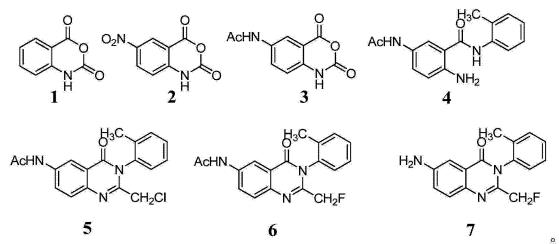
1)US3966731报道了以N- (2_氨基_5_硝基苯甲酰基)邻甲苯胺为原料,先与氟代乙酰氯缩合后再在醋酸酐作用下环合生成6-硝基-2-氟甲基-3- (2-甲基苯基)-4(3H)-1, 3- 二氮杂萘酮,最后在Pd-C催化作用下氢化还原得到氟喹酮。
该工艺总收率达66.6%,但是该工艺使用了氟代乙酰氯,该化合物沸点低,毒性大、腐蚀性大且价格昂贵,目前尚未产业化。
2)JP51105083报道了以N- (2_氨基`_5_硝基苯甲酰基)邻甲苯胺为原料,首先与氯乙酰氯直接环合生成2-氯甲基-3- (2-甲基苯基)-6-硝基-4 (3H)-喹唑啉酮,接着进行氟交换和还原反应生成最终产品氟喹酮。
该工艺总收率41.1%,该法用氯乙酰氯替代氟代乙酰氯,操作更安全,但是在对该工艺进行验证过程中发现关键的氟交换步骤硝基同时也能被氟取代,导致该步反应收率非常低,生产成本居高不下。
3)JP51105082报道了以N-(2_氨基-5-硝基苯甲酰基)邻甲苯胺为原料,先与氯乙酰氯环合,再进行硝基还原后上保护,接着进行氟交换和水解脱保护得到最终产品氟喹酮。
该工艺总收率28%,该法同样用氯乙酰氯替代氟乙酰氯,但不仅增加了氟交换反应还增加了上保护和脱保护反应,反应步骤多,总收率较低,成本高。对该工艺进行验证过程中发现在Pd-C通氢还原过程中发现脱氯现象,导致该步反应的收率非常低。如图4所示。
发明内容
本发明的目的在于提供一种反应条件温和、操作简便、产品收率高、生产成本低、绿色安全的氟喹酮的制备方法。
为实现上述目的,本发明采用的技术方案为:
一种氟喹酮的制备方法,包括如下步骤:
(1)、将靛红酸酐I溶于质量浓度为95%_98%的浓硫酸中,于0-10°C内缓慢滴加硝化试剂,滴完后保温反应0.5-2h,反应液缓慢倾倒入冰水混合物中,析出黄色固体,抽滤、水洗、真空干燥得5-硝基靛红酸酐2 ;所述靛红酸酐I与硝化试剂的投料物质的量比为1:1.1-1.3 ;
(2)、将5-硝基靛红酸酐2、有机溶剂A及雷尼镍加入高压釜中,通氢气维持釜内压力为0.5-2.0MPa,搅拌并升温至30-80°C反应0.5-4h,过滤回收雷尼镍,滤液中直接加入乙酸酐,室温下继续搅拌反应0.5-2h,减压浓缩回收溶剂,残留物经真空干燥得5-乙酰氨基靛红酸酐3 ;所述5-硝基靛红酸酐2与雷尼镍的投料质量比为1:0.03-0.08 ;5-硝基靛红酸酐2与乙酸酐的投料物质的量比为1:1.0-1.2 ;
(3)、将5-乙酰氨基靛红酸酐3溶于有机溶剂B中,加入邻甲苯胺,50-110°C内反应3-5h,减压浓缩回收溶剂和过量的邻甲苯胺,残留物经真空干燥得N- (2-氨基-5-乙酰氨基苯甲酰基)邻甲苯胺4 ;所述5-乙酰氨基靛红酸酐3与邻甲苯胺的投料物质的量比为1:1.0-1.5 ;
(4)、将N-(2_氨基-5-乙酰氨基苯甲酰基)邻甲苯胺4溶于冰醋酸中,室温搅拌下缓慢滴加氯乙酰氯,滴加完毕后,搅拌并升温回流反应1-5h,减压浓缩至干,残留物经真空干燥得6-乙酰氨基-2-氯甲基-3-( 2-甲基苯基)-4( 3H)-1, 3- 二氮杂萘酮5 ;所述N-( 2-氨基-5-乙酰氨基苯甲酰基)邻甲苯胺4与氯乙酰氯的投料物质的量比为1:0.9-1.1 ;
(5)、将6-乙酰氨基-2-氯甲基-3- (2-甲基苯基)-4 (3H)-1,3- 二氮杂萘酮5溶于有机溶剂C中,再加入现烘活化过的氟化钾,在季铵盐相转移催化剂的作用下搅拌回流反应1-5h,反应结束后经分离纯化得到6-乙酰氨基-2-氟甲基-3- (2-甲基苯基)-4(3H)-1, 3- 二氮杂萘酮6 ;所述6-乙酰氨基-2-氯甲基-3-(2-甲基苯基)-4(3H)-1,3- 二氮杂萘酮5与氟化钾、季铵盐相转移催化剂的投料物质的量比为1:3.0-5.0:0.1-0.2 ;
(6)、将6-乙酰氨基-2-氟甲基-3- (2-甲基苯基)-4 (3H)-1,3-二氮杂萘酮6投入到NaOH的乙醇-水混合溶液中,升温至40-80°C搅拌反应1-3h,反应结束后先减压回收乙醇,水层再用二氯甲烷萃取,有机层减压浓缩至干,所得残留物经异丙醇重结晶,真空干燥后得到6-氨基-2-氟甲基-3- (2-甲基苯基)-4 (3H)-1,3-二氮杂萘酮7,即氟喹酮。
本发明所述氟喹酮的制备方法步骤(1)中,所述浓硫酸的用量以靛红酸酐I的质量计为3-6mL/g ;所述硝化试剂选自浓硝酸或发烟硝酸;优选所述硝化试剂为65wt%-68wt%的浓硝酸。
本发明步骤(2)中,所述有机溶剂A选自乙腈、四氢呋喃、N,N_ 二甲基甲酰胺、N,N- 二甲基乙酰胺或它们任意比例的混合溶剂;优选所述有机溶剂A为四氢呋喃;所述有机溶剂A的加入量以5-硝基靛红酸酐2的质量计为10-15mL/g ;优选所述5-硝基靛红酸酐2与雷尼镍的投料质量比为1:0.03-0.05;优选5-硝基靛红酸酐2与乙酸酐的投料物质的量比为1:1.0-1.1。
本发明步骤(3)中,所述有机溶剂B选自乙腈、四氢呋喃、C1-C4的醇或它们任意比例的混合溶剂;优选所述有机溶剂B为乙醇;所述的机溶剂B的加入量以5-乙酰氨基靛红酸酐3的质量计为4-6mL/g ;优选所述5-乙酰氨基靛红酸酐3与邻甲苯胺的投料物质的量比为 1:1.1-1.3。
本发明步骤(4)中,所述冰醋酸的加入量以N- (2-氨基-5-乙酰氨基苯甲酰基)邻甲苯胺计4的质量计为4-6mL/g ;优选所述N- (2-氨基-5-乙酰氨基苯甲酰基)邻甲苯胺4与氯乙酰氯的投料物质的量比为1:0.95-1.05。
本发明步骤(5)中,所述有机溶剂C选自N,N_ 二甲基甲酰胺、硝基苯、环丁砜、乙腈、苯甲腈或它们任意比例的混合溶剂;优选所述有机溶剂C为乙腈;所述有机溶剂C的加入量以6-乙酰氨基-2-氯甲基-3- (2-甲基苯基)-4 (3H) -1, 3- 二氮杂萘酮5的质量计为4-6mL/g;所述季铵盐相转移催化剂选自苯基三甲基溴化铵、苯基三甲基氯化铵、苄基三乙基溴化铵、十六烷基三甲基溴化铵、四丁基溴化铵或四丁基氯化铵;优选所述季铵盐相转移催化剂为四丁基溴化铵;优选所述6-乙酰氨基-2-氯甲基-3- (2-甲基苯基)-4 (3H) -1, 3- 二氮杂萘酮5与氟化钾、季铵盐相转移催化剂的投料物质的量比为1:3.0-3.5:0.1-0.15。
本发明步骤(5)中 ,所述分离纯化的方法为:反应结束后反应液减压浓缩至干,加入二氯甲烷,过滤回收过量的氟化钾,滤液浓缩所得粗品用乙醇重结晶,真空干燥得6-乙酰氨基-2-氟甲基-3- (2-甲基苯基)-4 (3H) -1, 3- 二氮杂萘酮6。
本发明步骤(6)中,所述NaOH的乙醇-水混合溶液的溶剂是乙醇、水体积比配比1:0.4-0.6的混合溶液;所述NaOH的乙醇-水混合溶液中NaOH的质量浓度为4%-8% ;所述
6-乙酰氨基-2-氟甲基-3- (2-甲基苯基)-4 (3H)-1,3-二氮杂萘酮6与NaOH的投料物质的量比为1:1.5-2.0。
与现有技术相比,本发明的积极效果在于以价廉易得的靛红酸酐为起始原料合成关键中间体N- (2-氨基-5-乙酰氨基苯甲酰基)邻甲苯胺,大大降低了生产成本。革除了现有文献方法中高毒高害试剂氟代乙酰氯的使用,具有安全、环保的优点。在氟交换反应中使用四丁基溴化铵作为反应的相转移催化剂,大大提高了该步反应的转化率。由起始原料靛红酸酐制备目标产物氟喹酮的总收率可达60.0%以上。
具体实施方式:
以下以具体实施例用来进一步说明本发明的技术方案,但本发明的保护范围不限于此:
在100mL 二口烧瓶中,加入氢氧化钠(1.23g,30.8mol ),再加入7.50mL水和15.0mL乙醇使之溶解,最后加入6-乙酸氨基-2-氟甲基-3-(2-甲基苯基)-4(3H)-1, 3- 二氮杂萘酮6(5.00g, 15.4mmol)。70°C下搅拌2h后,先减压浓缩回收乙醇,水层再用二氯甲烷萃取(20mLX2)。合并有机层,减压浓缩至干得棕色黏稠油状物,加入25mL异丙醇重结晶得6-氨基-2-氟甲基-3- (2-甲基苯基)-4 (310-1,3-二氮杂萘酮7(3.7匕收率:85.0%)。m.p.194.2-195.7°C;HPLC 测量纯度为 99.0% (流动相为乙腈:20mmol/L KH2PO4 水溶液(pH3.25) =35:65,V/V)。