背景及概述[1][2]
急性髓系白血病(acutemyeloidleukemia,AML)是一组造血干细胞异质克隆性疾病,基因突变是除染色体异常之外,肿瘤细胞重要遗传学改变的特征。20%~30%患者存在Fms样的酪氨酸激酶-3(fms-liketyrosinekinase3,FLT3)基因内部串联重复序列(internaltandemduplication,FLT3-ITD)突变,也是AML发生率最高的一种基因改变。FLT3-ITD突变后,酪氨酸激酶的活性增高,导致自发激活,促进细胞增殖,与AML的发生、进展存在较密切关系,FLT3-ITD突变对AML的影响体现在长期预后,如:总生存期(overallsurvival,OS)、无病生存期(DFS)、无事件存活期(event-freesurvival,EFS)和更高的累计复发(CRR)的改变。midostaurin由瑞士诺华制药公司研发,译名为米哚妥林,代号PKC-412。
米哚妥林 (MS)是1977年从海洋链霉菌星形孢菌素培养液中分离的一种吲哚咔唑生物碱———十字孢碱(staurosporine)经结构改造获得的活性衍生物,中文俗称为4'-N-苯甲酰胺基-十字孢碱。米哚妥林是多重酪氨酸激酶受体的抑制药,美国食品药品管理局(FDA)于2016年2月19日授予米哚妥林与其他化学治疗(化疗)药联用,治疗新诊断为FLT3基因突变的AML成年患者突破性治疗药的地位,并给予快速通道审评的待遇。2017年4月28日获得美国FDA批准上市,商品名为Rydapt。此外,同时获批还可用于治疗成人侵袭性系统性肥大细胞增多(ASM)、伴有血液肿瘤的系统性肥大细胞增多症(SM-AHN)和肥大细胞白血病(MCL)等适应证。
非临床药理毒理学[1]
1. 致畸、致突变
尚未对米哚妥林进行致癌性研究。米哚妥林无基因毒性,体外细菌回复突变试验(Ames试验)或中国仓鼠V97细胞试验均为阴性。体外中国仓鼠卵巢细胞染色体畸变试验,米哚妥林能增加多倍体细胞的突变频率,但不引起染色体断裂。体内大鼠骨髓微核试验,给予耐受剂量(maximumtolerateddose,MTD)200mg·kg-1,按体表面积计算,1200mg·(m2)-1,相当于人用推荐剂量的20倍,不发生染色体断裂。
2. 对生殖能力的影响
分别对雄、雌性大鼠饲喂米哚妥林10,30和60mg·kg-1·d-1,按药物浓度-时间曲线下面积(AUC)计算,分别相当于人用推荐剂量的0.01,0.05和0.1倍,雄大鼠服药剂量≥10mg·kg-1·d-1时,观察到睾丸退行性变和萎缩,剂量为60mg·kg-1·d-1,精子计数减少,活动度降低和生殖器官质量降低;而观察到雌性大鼠胚胎再吸收增加,受孕率、植入物和活胚胎数降低。在一项对雄性犬进行为期3个月的毒理学研究,给予≥3mg·kg-1·d-1的剂量,相当于人用推荐剂量药物接触量的0.01倍,出现精子抑制作用。
临床药理毒理学[1]
1. 作用机制
米哚妥林是多重酪氨酸激酶受体的小分子抑制药。体外生化或细胞试验表明,米哚妥林及其人体内主要活性代谢物CGP62221和CGP52421有抑制野生型FLT3基因、FLT3-ITD突变和酪氨酸激酶结构域(tyrosinekinasedomain,TKD)突变、酪氨酸蛋白激酶KIT基因(tyrosine-proteinkinaseKit,KIT)的野生型和D816V突变体、血小板衍生生长因子受体α/β(plateletderivedgrowthfactorreceptor,PDGFRα/β)、血管内皮生长因子受体-2(vascularendothelialgrowthfactorreceptor,VEGFR-2)及丝氨酸/苏氨酸激酶蛋白激酶C(proteinkinaseC,PKC)族成员的活性。米哚妥林显示抑制FLT3基因受体信号和细胞增殖的能力,诱导白血病细胞表达ITD和TKD突变体的FLT3受体凋亡或过量表达野生型FLT3和血小板衍生生长因子(plateletderivedgrowthfactor,PDGF)受体细胞。米哚妥林也抑制肥大细胞KIT信号、细胞增殖和组胺释放的能力,并诱导细胞凋亡。
药效学[1]
1. 体外活性
米哚妥林及其代谢物CGP62221和CGP52421通过对酪氨酸激酶受体FLT3和KIT的抑制作用对于控制AML和SM病变起重大作用。抑制FLT3D835Y的半数抑制浓度(IC50,单位:nmol·L-1)分别为3.6及6.0,2.29和10.2;抑制FLT3的IC50分别为19.8,11.3和59.5,均能使白血病细胞繁殖周期停滞,表达细胞突变受体凋亡或过量表达野生型FLT3受体;抑制蛋白激酶Cn的IC50分别为160、无适用数据(notapplicable,NA)和NA;抑制KIT的IC50分别为330及600、NA和NA,能干扰KIT的异常信号,在表达KIT基因突变的BaF3细胞系和原发性肿瘤肥大细胞中,抑制肥大细胞的繁殖、生存和组胺释放;抑制脾脏酪氨酸激酶(Syk)的IC50分别为8.38,95及140,3.11和110;抑制集落刺激因子1受体(CSF1R)的IC50分别为25.9,NA和27.9;抑制血小板衍生的生长因子受体ɑ(PDGFRɑ)的IC50分别为34.2,NA和223;抑制PDGFRβ的IC50分别为35.2,NA和NA;抑制丝氨酸/苏氨酸的蛋白激酶Cɑ(proteinkinaseCɑ,PKCɑ)的IC50分别为203,22及280,152和927;抑制PKCβ1的IC50分别为263及30,NA和631;抑制PKCβ2的IC50分别为119及31,NA和NA;抑制PKC-g的IC50分别为24,NA和NA;抑制血管内皮生长因子(VEGF)受体KDR的IC50分别为86,NA和NA;抑制纤维母细胞生长因子受体-2(fibroblastgrowthfactorreceptor-2,FGFR-2)的IC50分别为91,未评估(NE)和NE;抑制原癌基因酪氨酸蛋白激酶(ROS1)的IC50分别为NA,22.9和NA。
2. 心脏电生理学
一项随机、安慰药、阳性药莫西沙星(moxifloxacin)对照,多剂量、盲态、平行组研究,评价口服米哚妥林75mg,bid,为期3d,对QTc间期延长的影响。结果表明,QTc间期延长或米哚妥林及其活性代谢物CGP62221和CGP52421的浓度与QTc间期变化之间的关系均无临床意义。研究持续时间似乎不足以评价代谢物CGP52421对QT/QTc间期的影响。汇总晚期SM患者的临床研究结果表明,有4.7%患者基线后QTcF间期>480ms,未发现有患者QTcF间期>500ms,另有6.3%患者与基线比较,QTcF间期>60ms。另一项对于AML患者的随机、安慰药对照的研究,QTcF间期>480ms,服药组与安慰药组分别为10.1%和5.7%;QTcF间期>500ms,分别为6.2%和2.6%;QTcF间期>60ms分别为18.4%和10.7%[1-3]。
药动学[1]
米哚妥林的药动学显示出依时性特征,开始服药的第1周,血浆药物谷浓度(Cmin)开始上升,约4周后,达到Cmin的最高值,其后,下降至稳态;活性代谢物CGP62221有类似的趋势;而另一个代谢物CGP52421恰好相反,血浆药物浓度持续增加,4周后达到稳态。进食时口服米哚妥林50或100mg,bid,米哚妥林及其代谢物CGP62221和CGP52421的Cmin最高值和稳态相似,但其Cmin和AUC0-168h的均值与SD分别仅为空腹服药的(38±7)%、(28±3)%和(22±5)%。
1. 吸收:空腹服药,达到血浆药物浓度峰值(Cmax)的时间(tmax)均值为服药后1h(1~3)h。进食标准餐(含脂肪50g、蛋白21g和碳水化合物18g,相当于1913.46J)和进食高脂餐(含脂肪66g、蛋白32g和碳水化合物64g,4216.31J)与空腹服药比较,米哚妥林的接触量用AUCinf表示,分别增加22%和59%。Cmax分别减少20%和27%。进食标准餐和高脂餐时服药,达到tmax的时间均延迟,中位tmax分别为2.5h及3.0h。
2. 分布:米哚妥林的表现分布容积几何估算均值与变异系数(CV)95.2L(31%)。米哚妥林及其代谢物主要分布于血浆。在体外,米哚妥林、CGP62221和CGP52421与血浆蛋白结合率>99.8%,米哚妥林主要与α1-酸性糖蛋白结合]。
3. 消除:米哚妥林终末端半衰期均值与CV为19h(20%),CGP62221为32h (31%),CGP52421为482h(25%)。健康受试者服米哚妥林50mg后,总清除率为3.4L·h。
4. 代谢:米哚妥林主要被CYP3A4代谢为2个活性代谢物:CGP62221为O-脱甲基化产物,CGP52421为单羟基化产物,其放射性均值与标准偏差(SD)分别占循环中放射性总量的(28±2.7)%和(38±6.6)%。
5. 排泄:95%的剂量从粪排泄,其中,91%为代谢物,4%为未变化原型药。尿中仅回收5%剂量。
6. 特殊人群药动学:20~94岁患者,轻至中度总胆红素升高,分别>正常上限(upperlimitofnormal,ULN)的1.0~1.5倍或1.5~3.0倍,轻至中度肝损伤患者,天门冬氨酸氨基转氨酶(AST)>ULN或任何数值,轻至中度肾损伤患者,肌酐清除率估算值(CLcr)≥30mL·min-1,对米哚妥林及其代谢物CGP62221和CGP52421的药动学无临床意义上的影响。尚不清楚严重肝损伤患者(总胆红素>3.0倍UL和AST为任何数值)和严重肾损伤患者(CLcr=15~29mL·min-1)对米哚妥林及其代谢物CGP62221或CGP52421药动学的影响。
不良反应
1. 不良反应概况:研发公司已完成23项临床试验研究,仅对2项Ⅱ期临床和1项Ⅲ期临床试验提供详尽的安全性数据,累计纳入患者859例,因试验条件各异,给药时间长短不一,临床发生的不良反应有较大差别,只按各批试验结果统计不良反应发生率。
2. AML的临床试验:纳入717例患者,完成临床试验研究后,可供评价的不良反应,治疗组(n=355),安慰药组(n=354)。按任何级别和3~5级不良反应发生率及两组用Fisher精确检验后的P值,顺次列举:血液学毒性,血小板减少,为97.5%(346/355),96.6%(342/354)和0.52;中性粒细胞减少95.2%(338/355),95.8% (339/354)和0.86;贫血92.7%(329/355),87.9%(311/354)和0.03;白细胞减少26.2% (93/355),29.7%(105/354)和0.32;淋巴细胞减少19.2%(68/355),22.0%(78/354)和0.35;其他血液或骨髓事件0.3%(1/355),1.1%(4/354)和0.22;骨髓细胞减少0%,0.3%(1/354)和0.50;非血液学毒性,发热性嗜中性粒细胞减少81.7%(290/355),82.5%(292/354)和0.84;感染52.4%(186/355),50.3%(178/354)和0.60;淋巴细胞减少19.2%(68/355),22.0%(78/354)和0.35;腹泻15.8%(56/355),15.3%(54/354)和0.92;低钾血症13.8%(49/355),16.9%(60/354)和0.25;疼痛13.2%(47/355),12.4% (44/354)和0.82;ALT升高12.7%(45/355),9.3%(33/354)和0.19;皮疹或脱皮14.1% (50/355),7.6%(27/354)和0.008;疲乏9.0%(32/355),10.5%(37/354)和0.53;肺炎或肺浸润7.9%(28/355),8.2%(29/354)和0.89;恶心5.6%(20/355),9.6%(34/354)和0.05;低钠血症8.7%(31/355),6.5%(23/354)和0.32;高胆红素血症7.0%(25/355),7.9%(28/354)和0.67;黏膜炎或口炎6.2%(22/355),7.9%(28/354)和0.38;低磷血症5.4%(19/355),8.2%(29/354)和0.14;低钙血症6.8%(24/355)、5.9%(21/354)和0.76。
3. 晚期SM的临床试验:纳入142例晚期SM患者,包括临床试验三116例意向治疗患者和试验四26例ASM患者,按发生所有级别和≥3级的不良反应发生率依次为如下。胃肠道紊乱:恶心为81.7%(116/142)和6.3%(9/142);呕吐68.3%(97/142和6.3%(9/142);腹泻54.2%(77/142)和7.7%(11/142);腹痛33.8%(48/142)和6.3% (9/142);便秘28.9%(41/142)和0.7%(1/142);胃肠道出血14.1%(20/142)和9.2% (13/142)。
一般性疾病:水肿40.1%(57/142)和7.0%(10/142);疲乏33.8%(48/142)和9.2%(13/142);发热26.8%(38/142)和4.2%(6/142)。感染:上呼吸道感染30.3%(43/142)和0.7%(1/142);泌尿道感染16.2%(23/142)和2.8%(4/142);肺炎9.9%(14/142)和7.7%(11/142);疱疹病毒感染9.9%(14/142)和0.7%(1/142)。肌肉骨骼和结缔组织病:肌肉骨骼疼痛35.2%(50/142)和4.2%(6/142);关节痛19.0%(27/142)2.1%(3/142)。
神经系统紊乱:头痛26.1%(37/142)和0.7%(1/142);眩晕12.7%(18/142)和0%。呼吸系统及胸部不适:呼吸困难23.2%(33/142)和7.0%(10/142);咳嗽18.3%(26/142)和0.7%(1/142);胸腔积液12.7%(18/142)和4.2%(6/142);鼻出血12.0%(17/142)和2.8%(4/142)。
其他不适:皮疹14.1%(20/142)和2.8%(4/142);QT间期延长11.3%(16/142)和0.7%(1/142);失眠11.3%(16/142)和0%;肾功能障碍11.3%(16/142)和4.9%(7/142)。实验室检测:淋巴细胞减少66.2%(94/142)和42.3%(60/142);白细胞减少61.3% (87/142)和19.0%(27/142);贫血59.9%(85/142)和38.0%(54/142);血小板减少50.0% (71/142)和26.8%(38/142);嗜中性粒细胞减少49.3%(70/142)和21.8%(31/142);高血糖80.3%(114/142)和18.3%(26/142);碱性磷酸酶升高38.7%(55/142)和9.2% ( 13/142);低钙血症38.7%(55/142)和2.1%(3/142);脂酶升高37.3%(53/142)和18.3%(26/142);高尿酸血症37.3%(53/142)和11.3%(16/142);谷氨酰转肽酶(GGT)升高35.2%(50/142)和9.2%(13/142);低钠血症33.8%(48/142)和4.9%(7/142);AST升高31.7%(45/142)和2.8%(4/142);ALT升高31.0%(44/142)和4.2%(6/142);高胆红素血症28.9%(41/142)和4.2%(6/142);低蛋白血症26.8%(38/142)和0.7%(1/142);低钾血症25.4%(36/142)和6.3%(9/142);肌酐升高25.4%(36/142)和0.7%(1/142);高钾血症23.2%(33/142)和4.2%(6/142);低磷血症21.8%(31/142)和0.7%(1/142);淀粉酶升高19.7%(28/142)和7.0%(10/·1336·HeraldofMedicineVol.36No.11November2017142);低镁血症19.7%(28/142)和0%。
适应证[1]
1. AML:米哚妥林适用于与阿糖胞苷及柔红霉素联用的标准诱导治疗及阿糖胞苷巩固化疗后,对新诊断为基因FLT3突变阳性成年AML患者的治疗,不适用作为AML患者的单药诱导治疗。
2. ASM:米哚妥林适用于ASM、SM-AHN和MCL成年患者的治疗。
剂量和服法[1]
1. 剂型与规格:Rydapt为软胶囊剂,只有一种规格,每粒含米哚妥林25mg。
2. 推荐剂量与服法
1)AML:患者在2个疗程的诱导期(第1~3天),静脉推注盐酸柔红霉素和第1~7天静脉滴注阿糖胞苷之后的第8~21天,与食物同服米哚妥林50mg,bid,一个疗程28d;其后是4个疗程的巩固治疗期,于第1天、第3天和第5天,每12h静脉滴注阿糖胞苷后的第8~21天,服米哚妥林50mg,bid;最后为维持治疗期,共12个疗程,服米哚妥林50mg,bid,直至疾病复发或不能耐受不良反应。
2)ASM、SM-AHN和MCL:患者与食物同服米哚妥林100mg,bid,一个疗程28d,直至疾病复发或不能耐受的不良反应。开始治疗的前4周,至少每周监测患者的毒性反应;其后8周,每隔一周监测一次;继续治疗,每个月监测一次。
3)ASM、SM-AHN和MCL:患者发生不良反应需要调整服药剂量①MCL患者的嗜中性粒细胞绝对计数(absoluteneutrophilcount,ANC)<1×109·L-1,不认为是米哚妥林的毒性。若患者ANC基线为(0.5~1.5)×109·L-1,ANC<0.5×109·L-1为服米哚妥林引起的毒性,应中断服药,直至ANC≥1×109·L-1,可在50mg,bid恢复服药,耐受性尚可,增加至100mgbid。停药后,ANC低值持续21d,应怀疑与米哚妥林有关。②血小板计数<50×109·L-1,不认为是毒性反应,患者的血小板计数基线为(25~75)×109·L-1,若<25×109·L-1,应中断服药,直≥25×109·L-1,可在50mg,bid,恢复服药,若耐受性尚可,增加至100mg,bid。停药后,血小板计数持续处于低值>21d,应怀疑与米哚妥林有关。③血红蛋白<8.0g·L-1,不认为是毒性反应或贫血会危及生命;而患者的血红蛋白基线为8.0~10g·L-1,血红蛋白<8.0g·L-1,应中断服药,直至血红蛋白≥8.0g·L-1,可在50mg,bid,恢复服药,若耐受性尚可,增加至100mgbid。停药后,血红蛋白持续处于低值>21d,应怀疑与米哚妥林有关。④尽管已采取抗呕吐治疗措施,仍发生3/4级恶心和(或)呕吐,可停药3d(6剂),再在50mg,bid恢复服药,若耐受性尚可,增加至100mg,bid。⑤发生非血液学3/4级不良反应,暂停服药,直至不良反应≤2级,恢复50mg,bid,若耐受性尚可,增加至100mg.
4)推荐服药方法:为减少服药后发生恶心和呕吐的风险,在服药前可预防性服止吐药:每天2次服药,约间隔12h;胶囊不可打开或压碎,应整粒吞服;缺失一剂或服药后呕吐,当日不可补足剂量,应在预定的时间表服下一次剂量;若米哚妥林与可能延长QT间期的药物同时服用,应采用心电图评估QT间期。
用药注意事项与警示[1]
1. 胚胎-胎儿毒性:根据作用机制和动物生殖研究的发现,妊娠期妇女服米哚妥林可能危害胎儿。动物研究表明,米哚妥林有致胚胎-胎仔毒性,包括后期致胚胎-胎仔死亡和降低胎仔出生后体质量,给予低于人用推荐剂量时,出现胎仔发育生长延迟。应忠告妊娠妇女对胎儿有潜在的风险。在米哚妥林治疗开始前7d内,应检验有生殖潜能女性的妊娠状态。忠告在米哚妥林治疗期间和末次剂量后至少4个月,必须使用有效避孕措施,女性的男伴侣在米哚妥林治疗期间和末次剂量后至少4个月应使用有效避孕措施。
2. 肺毒性患者:用米哚妥林单药治疗或与化疗联用治疗时,曾发生间质性肺病和肺炎,有一些是致命的病例。应监测患者的肺部症状。经受过间质性肺病或
肺炎的体征或症状后,没有查清其感染的病原学之前,应终止服用。
3. 儿童用药:尚未在儿童患者中确定米哚妥林的安全性和有效性,暂不推荐用药。
4. 老年患者用药:米哚妥林治疗晚期SM的临床研究在142例患者中,年龄≥65岁为45.1%(64/142),年龄≥75岁为11.3%(16/142)。受试者年龄≥65岁与较年轻受试者间比较,未观察到安全性或应答率有总体差别,但不排除老年患者个体有更大的敏感性。而治疗AML的临床研究中,没有包括足够数量年龄≥65岁的受试者,以确定这些患者是否与较年轻受试者有不同的反应。
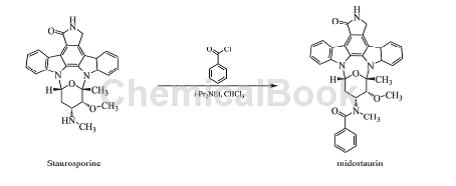
制备[3]
以星形孢菌素(Staurosporine)为原料,以二异丙基乙胺为缚酸剂,在氯仿溶液中以苯甲酰氯通过酰化反应制得。合成方法如图所示。
主要参考资料
[1] 治疗急性髓系白血病及系统性肥大细胞增生症新药-米哚妥林(midostaurin)
[2] CN200580028714.2米哚妥林用于治疗胃肠道基质瘤的用途
[3]米哚妥林