


How to synthesize Flumatinib mesylate?
Jan 19,2024
Description
Flumatinib mesylate, also known as HHGV-678, is a selective inhibitor of the tyrosine kinase BCR-ABL1. The drug was discovered and developed by Jiangsu Hengrui Pharmaceutical Group. It was approved by the CNMPA for the treatment of Philadelphia chromosome-positive chronic myeloid leukemia in the chronic phase (CML-CP). Structurally, flumatinib is similar to imatinib: the central phenyl ring was modified to a pyridine, and a CF3 group was appended to the benzamide group. With these changes, flumatinib demonstrated superior efficacy and a similar safety profile at 3, 6, and 12 months compared to imatinib during a Phase III clinical trial conducted in China.
Synthesis method
![BenzaMide, 4-[(4-Methyl-1-piperazinyl)Methyl]-N-[6-Methyl-5-[[4-(3-pyridinyl)-2-pyriMidinyl]aMino]-3-pyridinyl]-3-(trifluoroMethyl)-, MonoMethanesulfonate (9CI)](/NewsImg/2024-01-19/6384128129763282338190080.jpg "BenzaMide, 4-[(4-Methyl-1-piperazinyl)Methyl]-N-[6-Methyl-5-[[4-(3-pyridinyl)-2-pyriMidinyl]aMino]-3-pyridinyl]-3-(trifluoroMethyl)-, MonoMethanesulfonate (9CI)")
Although several strategies for synthesizing flumatinib have been reported, researchers at Hansoh Pharma recently published a patent describing a process route to the drug. Sulfonyl pyrimidine 278 and nitropyridine 279 underwent a substitution reaction to produce the aminopyrimidine core structure 280 in 83% yield. As an interesting alternative to Sandmeyer conditions, nitropyridine 280 was reacted with tri(p-chlorophenyl)phosphine and PhPOCl2 at elevated temperature to give chloride 281 in 97% yield. Flumatinib was then obtained in 94% yield from the copper-catalyzed coupling of 281 with amide 282 in the presence of base and tert-butyl hydroquinone (TBHQ)[1]. The final salt formation with methanesulfonic acid gave flumatinib mesylate in 98% yield.
Pharmacokinetic
Pharmacokinetic (PK) data of flumatinib showed that it was safe and well-tolerated, and Cmax and AUC0–t were linearly related to doses in the 200–1000 mg range. Preclinical pharmacokinetic studies showed that, in rats and beagles, the maximum blood concentration could be reached in about 5 h after oral administration of the drug. Furthermore, flumatinib mesylate was widely distributed in tissues, with tissue drug concentration higher than plasma concentration. The parent drug, flumatinib, was present in plasma, urine, and feces[2]. The primary metabolites in plasma were N-desmethyl flumatinib (M1), which was approximately 10% of that of the parent drug in plasma and has been shown to have similar pharmacological properties as the parent drug. The amide hydrolysis product (M3) was inactive but approximately 30% of that of the parent drug in plasma.
References
[1] Yun Kuang. “Effect of high-fat diet on the pharmacokinetics and safety of flumatinib in healthy Chinese subjects.” Cancer Chemotherapy and Pharmacology 86 3 (2020): 339–346.
[2] Andrew C. Flick. “Synthetic Approaches to the New Drugs Approved during 2019.” Journal of Medicinal Chemistry 64 7 (2021): 3604–3657.
- Related articles
- Related Qustion

Supplementation with pyridoxal 5'-phosphate monohydrate can synthesize neurotransmitters such as dopamine and serotonin, maintaining a healthy nervous system.....
Nov 4,2025Biochemical Engineering
Hydroxylamine sulfate induces methemoglobinemia, causing abnormal hemoglobin production. Mechanisms involve reactive oxygen species and free blood iron. Methylene blue may reduce methemoglobin levels.....
Jan 22,2024API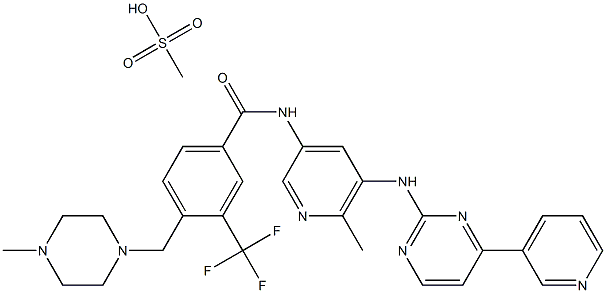
Flumatinib mesylate
895519-91-2You may like
Flumatinib mesylate manufacturers
- BenzaMide, 4-[(4-Methyl-1-piperazinyl)Methyl]-N-[6-Methyl-5-[[4-(3-pyridinyl)-2-pyriMidinyl]aMino]-3-pyridinyl]-3-(trifluoroMethyl)-, MonoMethanesulfonate (9CI)
-
![895519-91-2 BenzaMide, 4-[(4-Methyl-1-piperazinyl)Methyl]-N-[6-Methyl-5-[[4-(3-pyridinyl)-2-pyriMidinyl]aMino]-3-pyridinyl]-3-(trifluoroMethyl)-, MonoMethanesulfonate (9CI)](https://img.chemicalbook.com/ProductImageEN/2020-1/Small/e67b1ebe-50bc-46b6-8b85-a27e8b807a21.png)
- $1.00 / 1KG
- 2020-01-05
- CAS:895519-91-2
- Min. Order: 1KG
- Purity: 95--99%
- Supply Ability: 1ton