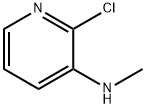
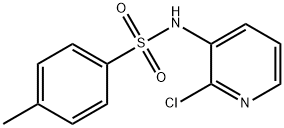
2-Chloro-3-methylaminopyridine synthesis
- Product Name:2-Chloro-3-methylaminopyridine
- CAS Number:40932-43-2
- Molecular formula:C6H7ClN2
- Molecular Weight:142.59
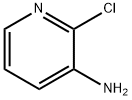
6298-19-7

74-88-4
40932-43-2
2-Chloro-3-aminopyridine (0.0465 mol) was dissolved in tetrahydrofuran (45 mL) under nitrogen protection and cooled to -78 °C. A 2.0 M solution of lithium diisopropylammonium (LDA, 0.0513 mol) was added slowly and dropwise. The reaction mixture was slowly warmed to 0 °C and stirred at this temperature for 1 hour. Subsequently, the reaction mixture was cooled again to -78 °C and iodomethane (0.0582 mol) was added. The reaction mixture was slowly warmed to room temperature and stirring was continued for 16 hours. Upon completion of the reaction, the reaction was quenched by the addition of saturated ammonium chloride solution and the mixture was extracted with ethyl acetate. The organic layers were combined, dried with anhydrous sodium sulfate, filtered and the solvent was concentrated under reduced pressure. The residue was purified by silica gel short column chromatography (eluent: hexane/ethyl acetate, 80/20). The fraction containing the target product was collected and the solvent was concentrated under reduced pressure to give 5.91 g of 2-chloro-3-methylaminopyridine (intermediate compound 1) in 89% yield.
6298-19-7
513 suppliers
$6.00/5g
74-88-4
357 suppliers
$15.00/10g
40932-43-2
89 suppliers
$24.00/100mg
Yield:40932-43-2 89%
Reaction Conditions:
Stage #1: 2-chloro-3-aminopyridinewith lithium diisopropyl amide in tetrahydrofuran at -78 - 0; for 1 h;
Stage #2: methyl iodide in tetrahydrofuran at -78 - 20; for 16 h;
Steps:
A.1.a A. Preparation of the intermediate compounds; Example A. 1; a. Preparation of intermediate compound 1
To a solution of 2-chloro-3-pyridinamine (0.0465 mol) in THF (45ml) at-78°C under N2 flow, LDA 2. 0M (0.0513 mol) was added dropwise. The mixture was allowed to warm to 0°C and was stirred for 1 hour and then cooled TO-78°C. Then CH3I (0.0582 mol) was added and the reaction mixture was allowed to warm to room temperature and was stirred for 16 hours. A saturated NH4CL-SOLUTION was added and the mixture was extracted with AcOEt. The separated organic layer was dried (NA2S04), filtered and the solvent was evaporated. The residue was purified by short open column chromatography over silica gel (eluent: hexane/AcOEt 80/20). The product fractions were collected and the solvent was evaporated. Yield: 5. 91g of intermediate compound 1 (89%).
References:
WO2004/106298,2004,A1 Location in patent:Page 19-20
209798-48-1
66 suppliers
$29.00/100mg
40932-43-2
89 suppliers
$24.00/100mg
6298-19-7
513 suppliers
$6.00/5g
40932-43-2
89 suppliers
$24.00/100mg
106320-35-8
0 suppliers
inquiry
40932-43-2
89 suppliers
$24.00/100mg
100062-14-4
0 suppliers
inquiry
40932-43-2
89 suppliers
$24.00/100mg