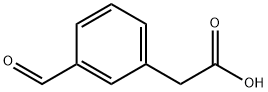
(3-ForMyl-phenyl)-acetic acid synthesis
- Product Name:(3-ForMyl-phenyl)-acetic acid
- CAS Number:34956-29-1
- Molecular formula:C9H8O3
- Molecular Weight:164.16
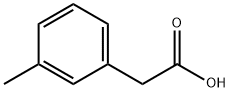
621-36-3
34956-29-1
General procedure for the synthesis of 3-formylphenylacetic acid from 3-methylphenylacetic acid: N-bromosuccinimide (2.37 g, 13.32 mmol) was added to a chloroform (30 mL) solution of 2-(m-tolyl)acetic acid. The reaction mixture was heated to reflux for 8 hours, followed by removal of the solvent under vacuum. The residue was dissolved in a solvent mixture of ethanol (30 mL) and water (30 mL) and hexamethylenetetramine (5 g, 35.7 mmol) was added. The mixture was heated to reflux and maintained for 4 hours. Under reflux conditions, concentrated hydrochloric acid (5.9 mL) was slowly added. Continue heating to reflux for 30 minutes, then cool to room temperature. Water (30 mL) and dichloromethane (30 mL) were added and the organic phase was separated and filtered through hydrophobic glassine. The solvent was removed under vacuum. The residue was dissolved in saturated aqueous sodium bicarbonate and washed with dichloromethane (2 x 15 mL). The aqueous phase was acidified with aqueous 2M hydrochloric acid to pH < 2. The acidified aqueous phase was extracted with dichloromethane (2 x 15 mL), the organic phases were combined and filtered through a hydrophobic glass. The solvent was removed in vacuo to afford 3-formylphenylacetic acid as an off-white solid (1.35 g, 62% yield). The product was characterized by 1H NMR (400 MHz, CDCl3): δ 10.01 (s, 1H), 7.84-7.79 (m, 2H), 7.60-7.48 (m, 2H), 3.75 (s, 2H).LCMS (Method 1): [MH+] = 165, retention time 2.77 min.
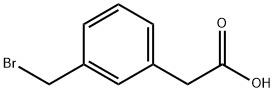
118647-53-3
45 suppliers
$60.00/1g
34956-29-1
48 suppliers
$44.00/100mg
Yield:34956-29-1 72%
Reaction Conditions:
Stage #1: (3-bromomethyl-phenyl)-acetic acidwith hexamethylenetetramine;water in ethanol; for 4 h;Reflux;Inert atmosphere;
Stage #2: with hydrogenchloride in ethanol;water; for 0.5 h;Reflux;Inert atmosphere;
Steps:
15 Step 2. 2-(3-formylphenyl)acetic acid (15.3)
Intermediate 15.2 (1 g,4.4 mmol) was dissolved in a 1: 1 mixture of EtOH (10 mL) and water (10 mL) and hexamethylentetramine (1.66 g,11.88 mmol) was added. The mixture was heated to reflux for 4 h. Concentrated HCl (2 mL) was added cautiously to the mixture at reflux. Stirring was continued at this temperature for an additional 30 min and then reaction mixture was allowed to cool. The solvent was removed in vacuo. The crude was taken up with water and the pH was adjusted to 8 by the addition of NaHC03. The aqueous phase was washed twice with EtOAc (2 x 20 mL). Then the pH was adjusted to 3 by the addition of 3N HCl. The crude mixture was extracted with EtOAc (3 x 20 ml). The organic phase was washed with brine,dried over Na2S04, filtered,and concentrated under reduced pressure. The title compound 15.3 (520 mg,3.2 mmol) was obtained as white solid. Yield 72%.XH NMR (200 MHz,CDCI3) δ 3.71 (s,2H), 7.49 (m,2H), 7.61 (s,2H), 10.03 (s,1H).
References:
WO2018/69532,2018,A1 Location in patent:Paragraph 0547; 0569
621-36-3
195 suppliers
$8.00/1g
34956-29-1
48 suppliers
$44.00/100mg

210115-24-5
2 suppliers
inquiry
34956-29-1
48 suppliers
$44.00/100mg
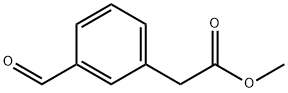
142327-44-4
40 suppliers
$99.00/100mg
34956-29-1
48 suppliers
$44.00/100mg
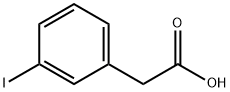
1878-69-9
178 suppliers
$27.00/1g
34956-29-1
48 suppliers
$44.00/100mg