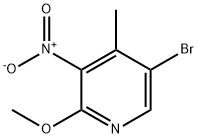
5-Bromo-2-methoxy-4-methyl-3-nitropyridine synthesis
- Product Name:5-Bromo-2-methoxy-4-methyl-3-nitropyridine
- CAS Number:884495-14-1
- Molecular formula:C7H7BrN2O3
- Molecular Weight:247.05
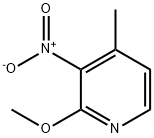
160590-36-3
884495-14-1
The general procedure for the synthesis of 5-bromo-2-methoxy-4-methyl-3-nitropyridine using 2-methoxy-3-nitro-4-methylpyridine as starting material was as follows: sodium acetate (365 g, 5.37 mol) was slowly added to a stirring solution of 2-methoxy-4-methyl-3-nitropyridine (250 g, 1.49 mol) in acetic acid (1.5 L) at room temperature. Subsequently, bromine (Br2, 639 g, 4.00 mol) was added dropwise and the dropwise addition time was controlled to be less than 30 minutes. After addition, the reaction mixture was warmed up to 80 °C for 12 h, during which the progress of the reaction was monitored by thin-layer chromatography (TLC) to confirm that the reaction was complete. Upon completion of the reaction, the mixture was cooled to 0 °C and 10% aqueous solution (1.5 L) and saturated aqueous sodium sulfate solution (1.5 L) were added sequentially to quench the reaction. The resulting solid product was collected by filtration, washed with water, and then dried under reduced pressure to finally obtain 5-bromo-2-methoxy-4-methyl-3-nitropyridine (302 g, 82.2% yield) as a light yellow solid. The structure of the product was confirmed by 1H NMR (400 MHz, DMSO-d6): δ 8.25 (s, 1H), 3.94 (s, 3H), 2.29 (s, 3H).
100367-40-6
239 suppliers
$10.00/1g
884495-14-1
190 suppliers
$18.00/1g
Yield:884495-14-1 70.44%
Reaction Conditions:
Stage #1:methanol with acetyl chloride at 0; for 0.166667 h;
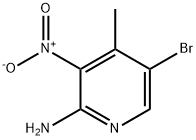
Stage #2:2-amino-3-nitro-4-methyl-5-bromopyridine with tert.-butylnitrite at 0 - 25; for 5 h;
Steps:
1A.B
Part B:; This reaction was conducted under inert gas protection. The reaction vessel was first charged with 2000 mL methanol and cooled to about 0° C. with slight agitation. Then 9.1 kg acetyl chloride was added. The exothermic reaction was then cooled and agitated for 10 minutes. The next addition was 100 g of 2-amino-5-bromo-3-nitro-4-picoline (the compound of formula 2) at 0° C. Then 236.5 g of t-butyl nitrite was added at a rate such that the temperature did not exceed 5° C. The slight evolution of nitrogen gas was noted. After the completion of the reaction, cooling was removed and the reaction mixture within the vessel was allowed to warm to 25° C. in about 30 minutes. The mixture was agitated at 25° C. for about 3-4 hours. After 4-5 hours a clear solution was obtained. Reaction completeness was monitored by HPLC after about 4 hours. The reaction was complete after about 5 hours. The reaction mixture was concentrated in vacuo to about 1000 mL. Then 500 mL of water was added and the product precipitated. Then 250 mL saturated sodium bicarbonate solution was added with good agitation to neutralize the HCl and dissolve the hydroxy impurity. The mixture was agitated at 20-25° C. for about 15 minutes and then the precipitate was collected and washed with 1000 mL of water. The product was then dried at 40° C. in vacuo. Yield was 75.0 g (70.44%), mp. 132° C. IR (KBr, cm-1): 1633, 1581, 1538, 1512, 1458, 1377, 1344, 1321, 1244, 869, 779. 1H-NMR (DMSO-d6) (δ, ppm): 2.31 (s, 3H), 3.96 (s, 3H), 8.55 (s, 1H): 13C-NMR (DMSO-d6) (δ, ppm): 17.49, 54.91, 99.41, 114.39, 141.02, 149.23, 153.46; HMS calcd for C7H7BrN2O3 245.96401 found (M+1): 246.97184; Elemental Analysis: calcd for C7H7BrN2O3: C, 34.03; H, 2.85; N, 11.34; found: C, 33.81; H, 2.91; N, 11.24.
References:
Bristol-Myers Squibb Company US2006/293304, 2006, A1 Location in patent:Page/Page column 29
160590-36-3
99 suppliers
$54.00/1g
884495-14-1
190 suppliers
$18.00/1g
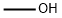
67-56-1
790 suppliers
$7.29/5ml-f
100367-40-6
239 suppliers
$10.00/1g
884495-14-1
190 suppliers
$18.00/1g
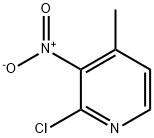
23056-39-5
332 suppliers
$6.00/1g
884495-14-1
190 suppliers
$18.00/1g
67-56-1
790 suppliers
$7.29/5ml-f
100367-40-6
239 suppliers
$10.00/1g
884495-14-1
190 suppliers
$18.00/1g
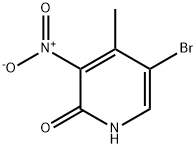
228410-90-0
123 suppliers
$10.00/250mg