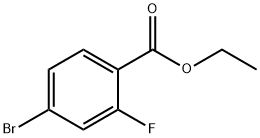
Ethyl 4-BroMo-2-fluorobenzoate synthesis
- Product Name:Ethyl 4-BroMo-2-fluorobenzoate
- CAS Number:474709-71-2
- Molecular formula:C9H8BrFO2
- Molecular Weight:247.06

64-17-5
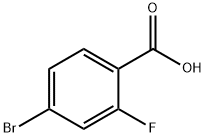
112704-79-7
474709-71-2
Step 1. Preparation of ethyl 4-bromo-2-fluorobenzoate To a stirred solution of 4-bromo-2-fluorobenzoic acid (1.50 g, 6.85 mmol) in dichloromethane (DCM, 26.0 mL) was added sequentially 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDCI, 1.60 g, 8.35 mmol), 4-dimethylaminopyridine (DMAP, 130 mg, 1.06 mmol) and anhydrous ethanol (EtOH, 2.00 mL). The reaction mixture was stirred at room temperature for 20 h and then washed with water and saturated saline in turn. The organic phase was dried over anhydrous sodium sulfate (Na2SO4), filtered and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (SiO2) with petroleum ether/ethyl acetate (1:5, v/v) as eluent to afford the target product ethyl 4-bromo-2-fluorobenzoate (1.52 g, 90% yield) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 1.39 (3H, t, J = 6.8 Hz), 4.39 (2H, q, J = 6.8 Hz), 7.32-7.37 (2H, m), 7.80-7.84 (1H, m).
64-17-5
823 suppliers
$10.00/50g
112704-79-7
538 suppliers
$15.00/5G
474709-71-2
77 suppliers
$23.00/1g
Yield:474709-71-2 90%
Reaction Conditions:
with N,N-dimethyl-4-aminopyridine;N-[3-(N,N-dimethylamino)-propyl]-N'-ethyl-carbodiimide hydrochloride in dichloromethane at 20; for 20 h;
Steps:
27.1
Step 1. Preparation of ethyl 4-bromo-2-fluorobenzoate To a stirred solution of 4-bromo-2-fluorobenzoic acid (1.50 g, 6.85 mmol) in DCM (26.0 mL) was added EDCI (1.60 g, 8.35 mmol), DMAP (130 mg, 1.06 mmol), and EtOH (2.00 mL) at room temperature. After being stirred for 20 hours at room temperature, the reaction mixture was washed with water and brine. The organic phase was dried over Na2SO4, filtered and concentrated in vacuo. The residue was purified by column chromatography on SiO2 (Hexanes/EtOAc=1/5) to give the desired product (1.52 g, 90%) as a colorless oil. 1H-NMR (400 MHz, CDCl3) δ 1.39 (3H, t, J=6.8 Hz), 4.39 (2H, q, J=6.8 Hz), 7.32-7.37 (2H, m), 7.80-7.84 (1H, m).
References:
US2012/53180,2012,A1 Location in patent:Page/Page column 29