【背景及概述】[1][2]
由睾酮在5α还原酶的作用下转变成的双氢睾酮(DH T)作用于前列腺组织,可导致前列腺组织增生。临床上已应用5α还原酶抑制剂来阻断这个环节,以有效地治疗前列腺增生症(BPH)。最近的研究发现,人体中5α还原酶有两种同工酶,即5α还原酶-1 和5α还原酶-2。5α还原酶-1 存在于身体任何有5α还原酶表达的部位,包括皮肤、肝脏、脂肪的腺体、大多数毛囊和前列腺。5α还原酶-2主要存在于前列腺及其他生殖组织、生殖器的皮肤、胡须和头皮毛囊,并与男胎儿的男性化有关。
在鼠的研究中发现,虽然两种同工酶对合成代谢和分解代谢都有作用,但1-型酶主要对雄激素和其他类固醇激素的分解代谢起作用,而2-型酶则主要对雄激素的合成代谢起作用。在正常前列腺组织、BPH 病人和前列腺癌病人的前列腺的所有区域(包括周围带、移行带和中心带)都有两种5α还原酶的mRNA ,但是在前列腺癌组织中只有5α还原酶-1 的表达增加。
非那雄胺是5α还原酶-2 的抑制剂,它在临床应用的剂量时只能抑制5α还原酶-2。为细胞内酶。分子式为C27H3OF6N2O2,分子量为528.5。度他雄胺为白色至淡黄色粉末,熔点为242~250℃,不溶于水,溶于乙醇(44mg· mL-1),甲醇(64 mg· mL-1),聚乙二醇400(3 mg· mL-1)度他雄胺(dutasteride)是一种新的5α还原酶的双重抑制剂,它既能抑制5α还原酶-1,也能抑制5α还原酶-2。它比非那雄胺更能使DHT 的浓度降低(94 .7 %对70 .8 %)。度他雄胺对5α还原酶-1 的抑制作用是非那雄胺的60 倍。服用度他雄胺后27 个月,前列腺癌的发病率比安慰剂组低50 %。
【适应症】[3]
治疗良性前列腺增生症(BPH)的中、重度症状。用于中、重度症状的良性前列腺增生症患者,降低急性尿潴留(AUR)和手术的风险。
【规格】[3]
胶囊:0.5mg。
【用法用量】[1]
推荐剂量为每日口服1次,1次0.5 g。虽然半衰期长,试验显示不会导致积蓄中毒。肾功能不良及老年患者不需调整剂量。肝功能不良的剂量调整研究因数据不足尚未建立。但由于度他雄胺经肝脏代谢,可能应减少用药剂量。
【药理作用】[1]
症状性良性前列腺增生(BPH)是男性老年患者的常见病,BPH可引起不同程度的膀胱输出受阻,并因排尿不畅和膀胱快速充盈而导致进展性尿频和尿急,患者夜间遗尿的发生率增加。病因主要是体内二氢睾酮(DHT)水平的升高。睾酮在5α还原酶的作用下转化成DHT,5α还原酶有Ⅰ 型和Ⅱ 型两种同工酶,Ⅱ 型同工酶对生殖系统发挥作用,Ⅰ 型同工酶主要作用于皮肤和肝脏的睾酮转化。度他雄胺同时抑制Ⅰ 型和Ⅱ 型5α还原酶,从而抑制睾酮转化成二氢睾酮,它与5α还原酶组成稳定的结合体,体内和体外试验显示结合体的离解非常慢,而且度他雄胺对雄激素受体没有亲和力。
【药代动力学】[1]
度他雄胺口服吸收较好,也可通过皮肤吸收,11.5%进入精液。口服1~2周血药浓度达峰值,99%与血浆蛋白结合,其中96.6%与α-糖蛋白结合,口服生物利用度不受食物影响。大部分通过肝脏代谢,代谢物经粪便排出。稳态时度他雄胺的消除半衰期为5 周,口服度他雄胺0.5mg· d-1 1 a的平均稳态浓度为40ng/mL,服药1个月的血药浓度为稳态浓度的65%,服用3个月约为稳态浓度的90%。疗程为6~12个月。
由于其半衰期长,停药4~6周仍可在血浆中检测到度他雄胺(>0.1 ng· mL-1)。度他雄胺能显著降低头皮、血浆中DHT水平,其降低DHT的药理效应呈剂量依赖性,每日口服度他雄胺0.5 mg,在用药1,2周测定DHT的血浆浓度,结果平均血浆浓度分别下降85%和90%,而治疗2周的患者,DHT平均血浆浓度下降94%,睾酮血浆浓度增加19%,仍然在生理范围内。同时促甲状腺激素在52周上升12.4%,促黄体激素增加到12%,12周增加到19%。
【不良反应】[2]
尽管度他雄胺在前列腺增生症和前列腺癌的治疗中有明显的作用,但是影响其临床应用的关键在于其副作用。与非那雄胺一样,其最常见的副作用有:耳鼻喉感染、肌肉骨骼疼痛上呼吸道感染。药物相关的副作用占19 %。勃起功能障碍、性欲减退、射精疾病、男性乳房发育。
在24 个月时,1 .1 %出现前列腺癌(安慰剂组为1 .9 %)。与性功能有关的副作用都是一时性的,主要出现在试验的早期,随后逐渐减少。在服药的第4 年,勃起功能障碍、性欲降低、射精疾病的发病率小于0 .5 %。男性乳房发育仅少数病人出现。总的说来,有关性的副作用是低的,其临床意义表现在只有极少数病人会因此而退出试验。未见有肝功能损害的报告。由于5α还原酶-1 存在于肝脏,这一点就有特殊的意义。
【药物相互作用】
度他雄胺在肝脏通过CYP3A4 代谢,体外试验显示,CYP3A4抑制药如利托那韦、酮康唑、维拉帕米、地尔硫、西咪替丁和环丙沙星可增高血浆中度他雄胺的浓度,而和坦索新、特拉唑嗪、华法林和考来烯胺没有药理学和药动学的相互作用。
【注意事项】[1]
妇女、儿童、对度他雄胺或其他5α还原酶抑制药易产生变态反应者禁用。因度他雄胺可致男性胎儿畸形,且可通过皮肤吸收,孕妇及可能怀孕的妇女应避免接触。服用度他雄胺的患者至少在停药6个月后方可献血。
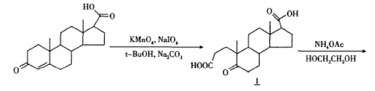
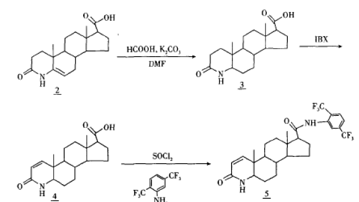
【制备】 [4]
以孕甾烯酮酸为起始原料,首先将A环开环,以乙酸铵代替氨气,引人N-杂原子关环,降低了对反应设备的要求。使用甲酸,DMF体系还原5位双键,避免使用H2还原。采用IBX进行脱氢,避开了剧毒的硒化合物,降低了对环境的污染,使合成更适用于工业生产。
【主要参考资料】
[1] 顾国良, 袁峰. 度他雄胺的药理作用与临床应用[J]. 医药导报, 2005, 24(3): 214-215.
[2] 何家扬. 度他雄胺治疗前列腺增生症和前列腺癌[J]. 現代泌尿外科雜誌, 2006, 11(1): 1-3.
[3] 度他雄胺软胶囊
[4] 何明华, 廖清江. 度他雄胺的合成[J]. 化工時刊, 2009, 23(3): 44-46.