背景及概述[1]
2-氯-3-硝基-6-甲基吡啶是一种有机中间体,可由2-氨基-6-甲基吡啶为原料先硝化得到2-氨基-3-硝基-6-甲基吡啶,然后重氮化反应得到2-羟基-3-硝基-6-甲基吡啶,最后氯代得到2-氯-3-硝基-6-甲基吡啶。2-氯-3-硝基-6-甲基吡啶可用于制备2-苯基吡啶与2-(4-二苯并呋喃)-吡啶。

制备[1][3]
报道一、
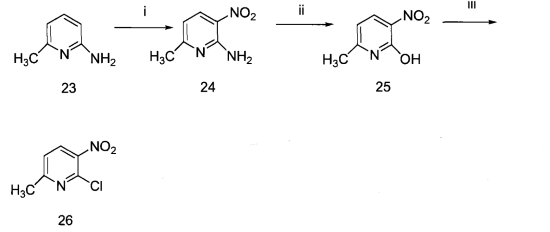
化合物24的合成
将浓硫酸(100ml)在冰浴下冷却,缓慢加入原料23(30g,0.28mol),冷却至0℃,缓慢加入体积比为1∶1的浓硫酸(98%)和浓硝酸(72%)混酸42mL,在0℃下反应1h.,放置12h。将反应液倒入2L冰水中,加浓氨水调节pH=7,过滤,滤饼干燥,得粗产品54g。将以上混合物经水蒸气蒸馏,得亮黄色液体,经乙酸乙酯萃取,在乙酸乙酯中重结晶,得产品12.5g,熔点:156.5-158.5℃(乙酸乙酯),收率:29%。
化合物25的合成
将化合物24(10g,0.065mol)加入100mL水中,搅拌下缓慢加入浓硫酸(12mL),冰浴冷却至0℃。分批加入亚硝酸钠(6.9g,0.098mol),在0℃下反应4h,放置12h。有大量黄色沉淀析出,减压抽滤,真空干燥,得黄色产品7.7g,收率77%。熔点:216.5-218.5℃(水)。
化合物26的合成
将化合物25(10g,0.065mol)加入50mL三氯氧磷中加热回流4h,减压蒸馏除去大部分的三氯氧磷,将其倒入200g冰水中,搅拌2h.,有大量沉淀析出,减压抽滤,真空干燥,得白色固体产品10g,收率89%。熔点:68.5-70.5℃(水)[J.O.C.,1955,20,1729-1731]。
报道二、
2-羟基-3-硝基-6-甲基吡啶(I-a)
将300mL浓硫酸缓慢加入含有152克(1.4mol)2-羟基-6-甲基吡啶的反应瓶,加毕,室温 滴加92mL(1.47mol)发烟硝酸。控制滴加速度,使内温低于60℃。滴毕,室温搅拌1hr。反应 液倒入冰水中,析出固体,过滤。滤液用碳酸钠调pH值至5~6,析出固体,抽滤。固体用 无水乙醇洗涤,水相用乙酸乙酯萃取,合并有机相,减压回收溶剂。干燥后得黄色固体120 克(56%)。(未分离,直接投下一步反应)
2-氯-3-硝基-6-甲基吡啶(I-b)
将上述混合物120克(0.8mol)、400mL三氯氧磷和200mL甲苯加入反应瓶中,机械搅拌, 升温至回流。TLC跟踪,6hrs后原料消失。减压回收过量的三氯氧磷和甲苯,残余膏状物倒 入冰水中,氢氧化钠水溶液调pH值至8~9。乙酸乙酯200mL×5萃取,合并有机相,减压回 收溶剂。粗品经柱层析(展开剂:乙酸乙酯∶石油醚=1∶40)得2-氯-3-硝基-6-甲基吡啶淡黄色 固体45克(收率:33%),mp:58~60℃。
1HNMR(300MHz,CDCl3):2.51(3H,s,-CH3),7.80~7.81(1H,m,ArH),8.92~8.96(1H,m,ArH),)
应用[2]
2-苯基吡啶与2-(4-二苯并呋喃)-吡啶为配体的铱杂配合物是广泛应用的有机磷光材料。二苯并呋喃结构扩展了配体的共轭平面,降低配合物的LUMO轨道能级,导致材料的发光红移,降低了不饱和绿光成分。2-氯-3-硝基-6-甲基吡啶制备2-苯基吡啶与2-(4-二苯并呋喃)-吡啶的方法如下:
(1)2-(2-甲氧基苯氧基)-6-甲基-3-硝基吡啶的合成
在装有回流冷凝管和机械搅拌的50L玻璃反应釜中,加入3Kg 2-氯-3-硝基-6-甲基吡啶、2.30Kg愈创木酚和2.34Kg DABCO,油浴加热温度设定为130℃反应3-4h,取样用液相色谱检测手段检测原料2-氯-3-硝基-6-甲基吡啶出峰完全消失时,即可停止加热;当油浴温度降低至80℃时,加入30L的1,2-二氯乙烷搅拌分散,继续降至室温,然后过滤,将滤饼用5L的1,2-二氯乙烷浸泡并抽绿,将水加入所得滤饼中搅拌洗涤15分钟,停止搅拌,进行分液;有机相先用硅胶柱脱色,再旋干回收1,2-二氯乙烷,得到黄色油状物,在所得黄色油状物中加入10L石油醚打浆,分散,抽滤,干燥得到淡黄色固体4.01Kg,该淡黄色固体经1H-NMR和13C-NMR确定结构为2-(2-甲氧基苯氧基)-6-甲基-3-硝基吡啶,液相色谱仪分析纯度为98%,收率为89%。
(2)2-(2-甲氧基苯氧基)-6-甲基-3-氨基吡啶的合成
在装有回流冷凝管和机械搅拌的50L玻璃反应釜中,加入3Kg上述步骤(1)中制备的2-(2-甲氧基苯氧基)-6-甲基-3-硝基吡啶,150g含钯量10%的湿钯碳,30L无水乙醇,搅拌下加热至70℃;用滴液漏斗滴加1.5L水合肼,反应2小时后,停止加热,回温至室温,过滤回收钯碳,混合液将乙醇旋干,得到淡黄色油状物,在所得淡黄色油状物中加入12L石油醚打浆,分散,抽滤,然后真空干燥得到类白色固体2.24Kg,该类白色固体经1H-NMR和13C-NMR确定结构为2-(2-甲氧基苯氧基)-6-甲基-3-氨基吡啶,液相色谱仪分析纯度为98%,收率为84%。
(3)2-甲基-8-甲氧基苯并呋喃[2,3-b]吡啶的合成
在50L双层低温釜中加入上述步骤(2)中制备的2-(2-甲氧基苯氧基)-6-甲基-3-氨基吡啶1.5Kg,碘化亚铜75g,二甲基亚砜30L,油浴加热至80℃,缓慢滴加亚硝酸异戊酯920g,加完后继续保温至80℃反应,取样采用液相色谱检测手段检测原料2-(2-甲氧基苯氧基)-6-甲基-3-氨基吡啶出峰完全消失时,将反应液加入60L水中,再加入25L石油醚,5L乙酸乙酯,充分搅拌,分离出水相,将有机相先用水洗涤,再用饱和氯化钠溶液洗涤,然后用硅胶柱脱色,将透过液旋干,得到白色固体0.95Kg,所得白色固体经1H-NMR和13C-NMR确定结构为2-甲基-8-甲氧基苯并呋喃[2,3-b]吡啶,收率63%,液相色谱仪分析产物纯度为99%。
参考文献
[1] [中国发明,中国发明授权] CN200810173259.0 硝基吡啶乙烯亚胺化合物、其药物组合物及其制备方法和用途
[2] [中国发明] CN201910341401.6 一种2-甲基-8-甲氧基苯并呋喃[2,3-b]吡啶的合成方法
[3] [中国发明,中国发明授权] CN200810018702.7 端粒酶抑制剂及其制备方法和用途