背景及概述[1][2]
对甲氧基苯乙酸(4-methoxyphenylacetic acid),通常为白色片状晶体,熔点为84-86℃,易溶于热水、乙酸乙酯、乙醇、乙醚等,可溶于冷水。对甲氧基苯乙酸是合成新一代抗抑郁药文发拉新(Venlafaxine)的中间体,也是合成葛根素、异黄酮类等多种心血管药物的关键性中间体。关于它的合成方法,国内外的报道主要有以下几种:a)氰化法,一般用对甲氧基苄氯和氰化钠反应生成对甲氧基苯乙腈,后者经水解得到对甲氧基苯乙酸,这种方法要使用剧毒的氰化物,因而存在严重的安全隐患;b)苯乙酮重排法,该法常以对甲氧基苯乙酮原料,与硫磺和六水哌嗪反应,经过重排及水解得到对甲氧基苯乙酸,但该法生成的硫化氢剧毒奇臭,收率也不高;c)羰基合成法,该法以对甲氧基氯苄为原料,在催化剂作用下,与CO 反应生成对甲氧基苯乙酸,这种方法的缺点是所使用的催化剂价格昂贵,CO 危害性也较大。
结构[2-5]
对甲氧基苯乙酸是一种重要的医药中间体,广泛地应用于医药、染料等方面。 例如对甲氧基苯乙酸是合成右美沙芬中间体1-(4-甲氧基苄基)-1,2,3,4,5,6,7,8-八氢异喹啉的重要原料;也是合成新一代抗抑郁药物文拉发新的重要原料;同时也是合成抗肿瘤、抗真菌及抗细菌药物菲并联啶类生物碱的重要中间体α,β-二芳基取代丙烯酸的重要原料;另外,对甲氧基苯乙酸也是合成葛根素、异黄酮类等多种心血管药物的关键性中间体。其他应用如下:
1. 制备酒石酸左洛啡烷。该方法以对甲氧基苯乙酸和2-(环己烯基)乙胺为起始原料经酰化缩合、Bischler-Napieralski成环反应、亚胺还原、醚键水解、拆分、N-烷基化、Grewe环化反应及成盐共九步反应。此方法以高收率、高纯度得到酒石酸左洛啡烷及各中间体,可作为工业化方法进行大规模生产。
2. 制备刺芒柄花素。先缩合,在干燥反应器中,加入间苯二酚、对甲氧基苯乙酸、三氟化硼乙醚,加热反应,反应毕,加入水,加热回流,冷却析出结晶,过滤,水洗至中性,干燥,得中间体;再环合,在干燥反应罐中,加入中间体、原甲酸三乙酯、吗啉、DMF和冰乙酸,分出反应付产物乙醇,加热回流,反应毕,减压回收DMF试剂,冷却,向剩余物中加入饱和碳酸氢钠溶液回流,降温,析出结晶,常规处理得粗品刺芒柄花素,经甲醇精致得到精品刺芒柄花素,本发明解决目前刺芒柄花素资源缺乏的问题,具有原料易得、反应高效、方便的优点,有很高的应用价值。
3. 制备2,4-二羟基-4-甲氧基脱氧安息香的制备方法,包括如下步骤:(1)、干燥容器中加入间苯二酚、氯化锌、对甲氧基苯乙酸,抽真空至-0.06Mpa以内,于120℃~140℃条件下搅拌反应30~120min;(2)、反应结束后,降温至100℃以下,加水溶解化料,搅拌30~100min,降温至5~30℃,结晶;(3)、过滤,滤饼用水洗涤至中性,干燥即得产物。本发明设计合理,使用氯化锌为催化剂,经过调节温度及压力,即反应条件温和在负压条件下,就可制备出刺芒柄花素的关键中间体,不需要特殊设备,对设备腐蚀性小,安全速度快,操作简便,废液小,污染小。
制备 [1,6]
方法1:以苯甲醚(2)为原料,在A lCl3 催化下与草酰氯单乙酯发生Friedel-C raft s 酰化反应生成对甲氧基苯乙酮酸乙酯(3),后者不经分离直接与水合肼发生Wolff-Kishner-Huang 反应得到目标产物(1),总产率为55 .4 %。本合成路线所使用的原料廉价,无需使用昂贵及剧毒试剂,工业前景较好。具体的反应式如图所示。

步骤1:对甲氧基苯乙酮酸乙酯(3)的合成
在装有电动搅拌和尾气吸收装置的250 mL 三口烧瓶中加入14 .7 g(0.11 mol)无水三氯化铝、120 mL1,2-二氯乙烷和10 .8 mL(0.10 mol)苯甲醚,搅拌均匀并且加热至45 ℃,加入12 .3 mL(0 .11 mol)草酰氯单乙酯,保温反应8 h。反应结束后冰水浴冷却,加入浓度为10 %的稀盐酸水溶液100 mL,分出有机层。依次经5 %碳酸氢钠溶液(50 mL ×2)和水(80 mL ×2)洗涤,无水硫酸钠干燥后过滤,滤液减压浓缩至干得到粗产品(3)17 .8 g,产率为85 .5 %。本品不经纯化,直接用于下一步的合成。
步骤2:对甲氧基苯乙酸(1)的合成
在250 mL 三口烧瓶中加入粗产品(3)17 .8 g(0 .086 mol)、110 mL 乙二醇以及12 mL(0 .24 mol)85 %的水合肼,缓慢升温至130 ℃反应2 h,然后将反应液冷至室温后加入13 .4 g(0 .24 mol)固体氢氧化钾,升温至180 ℃反应4 h,边升温边蒸出低沸物(约2 h),再回流反应4 h 。反应结束后冷却至室温,加入6 mol/L 的盐酸(约55 mL)调至pH 1~2 。加入水180 mL ,用乙酸乙酯萃取(50 mL ×2),合并有机相。合并后的有机相用水(10 mL ×2)洗涤,经无水硫酸钠干燥后过滤,滤液减压浓缩至干,剩余物用正己烷(62 mL)重结晶以除去邻位产物及其它杂质,抽滤并烘干滤饼后得到银白色鳞片状晶体(1)9.2 g ,此步产率为64 .4 %m .p .84~ 86 ℃。
方法2:一种以苯甲醚合成对甲氧 基苯甲酸方法,其反应式和操作步骤如下:
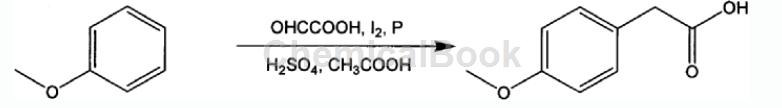
具有如下操作步骤:
1)将如下物料按照1mol苯甲醚、0.3-1.5mol乙醛酸、1-20mL浓酸、碘、红磷、100-1500mL冰醋酸的配方,依次加入到圆底烧瓶中;
2)在50-100℃条件下将上述物料搅拌2-12小时,并利用高效液相色谱 HPLC取样监测,待反应完毕后降至室温,滤掉红磷,收集滤液;
3)向上述滤液中加入10-100mL10%的乙酸钠水溶液,搅拌后,滴加浓盐酸调节混合液pH值至1-2,待析出沉淀,过滤,收集滤渣;
4)将上述滤液干燥,得到对甲氧基苯乙酸粗品,再经减压蒸馏得到纯品对甲氧基苯乙酸;
主要参考资料
[1] 对甲氧基苯乙酸的合成新工艺研究
[2] CN201510111704.0 对甲氧基苯乙酸的制备方法
[3] CN201610886326.8一种酒石酸左洛啡烷的制备方法
[4] CN201010600926.6一种刺芒柄花素的合成方法
[5] CN201610677984.62,4-二羟基-4-甲氧基脱氧安息香的制备方法
[6] CN201310319288.4一种由苯甲醚合成对甲氧基苯乙酸的方法