背景[1]
舒尼替尼由下式表示,其化学命名为N-[2-(二乙基氨基)乙基]-5-[(Z)-(5-氟-2-氧代-1,2-二氢-3H-吲哚-3-亚基)甲基)-2,4-二甲基-1H-吡咯-3-甲酰胺,是一种靶向和阻断多重选择的受体酪氨酸激酶(RTK)信号通路的口服酪氨酸激酶抑制剂(TKI)。
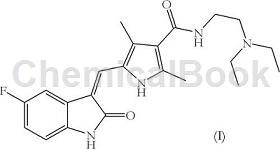
舒尼替尼
通过竞争性抑制ATP结合位点,舒尼替尼抑制一组密切相关的RTK的TK活性,所有这些RTK都与各种人恶性肿瘤有关:血管内皮生长因子受体(VEGFR-1,-2,-3)、血小板衍生生长因子受体(PDGF-R)、干细胞因子(KIT)、CSF-1R、Flt3和RET。因此,舒尼替尼适用于治疗癌症和肿瘤。目前,市售的舒尼替尼用于治疗不可切除的和/或转移性恶性胃肠道间质肿瘤(GIST)和晚期和/或转移性肾细胞癌(MRCC)。这种产品作为苹果酸舒尼替尼以专利药品名索坦进行销售。
应用[3]
舒尼替尼(Sunitinib)是由Sugen公司和辉瑞公司开发研制的一种口服、多靶点的受体酪氨酸激酶(Receptor tyrosine kinases,RTKs)抑制剂,而且是目前已知的作用靶点最多的靶向抗肿瘤药物之一,能对包括血管内皮生长因子受体(VEGFR)1、2、3,血小板生长因子受体(PDG-FR)α,β,KIT,FLT3和RET在内的等多个受体RTKs有较强抑制活性。这些RTKs均为跨膜蛋白,属于III和V类裂解激酶域家族成员。临床上常用于治疗标准治疗无效或不能耐受的恶性胃肠道间质瘤或转移性肾细胞癌。
它能抑制体内多种受体酪氨酸激酶的磷酸化,通过抑制生长和阻断肿瘤的血供,使肿瘤失去继续分裂和生长能力来对抗肿瘤,对于肿瘤生长、肿瘤细胞表达和肿瘤转移显示出较强的抑制作用。临床前研究表明,舒尼替尼显示出广谱抗肿瘤活性,包括小细胞肺癌,乳腺癌、结肠癌、黑色素瘤、神经交质瘤、肾细胞癌、表皮样癌和急性髓系白血病等。
乳腺癌中与多西紫杉醇、氟尿嘧啶和蒽环类药物联合、在小细胞肺癌中与顺铂联合以及在结直肠癌中与伊立替康联合可以增强抗肿瘤效应。临床试验表明,该药能延缓胃肠道间质肿瘤的生长速度,并能缩小肾细胞肿瘤的尺寸。在化疗或放疗的同时加用舒尼替尼有助于增强化疗和放疗的抗癌作用,因此在临床上有很好的应用前景。另外,它对乳腺癌、结肠直肠癌、前列腺癌和非小细胞肺癌的临床试验正在进行中。
作用机制及临床前研究
受体酪氨酸激激酶(RTKs)在一系列细胞生活周期(例如细胞生长、分化和死亡)的信号转导途径中起重要的作用。很多类型的肿瘤因为存在RTKs功能紊乱,而诱导肿瘤细胞生长、增殖及分化等。血管内皮生长因子受体1、2、3(VEGFR—l,2,3)具有酪氨酸激酶活性,能激活细胞内信号通路,最终引起肿瘤血管生成。血小板衍生生长因子受体a和B(PDGFR—Q,B)在很多肿瘤中均有过度表达。在肿瘤增殖过程中,PDGFR上调,刺激内皮细胞周围的问质细胞和成纤维细胞的生长和增殖。
舒尼替尼可以同时阻断VEGFR和PDGFR信号转导通路,从而达到更强的抗血管生成作用。此外,舒尼替尼还可以抑制其它RTKs,如碱性成纤维细胞因子(bFGF)、胎肝激酶3(Flt3)和干细胞因子受体(c—Kit)等。研究发现,同时阻断VEGFR、PDGFR及其它多种酪氨酸激酶的活性比单独阻断一种酪氨酸蛋白激酶的抗肿瘤效果更强。
在临床前研究中,舒尼替尼在很多细胞株和动物移植瘤模型中表现出抑制肿瘤生长和增殖的作用。在对HT一29和Col0205结肠癌、NcI一汜26非小细胞肺癌、W}1-266—4黑色素瘤、786-0肾细胞癌、A431表皮样癌移植瘤动物模型的研究中,每日给药一次能使肿瘤消退,而且不产生耐药现象。
在移植瘤动物模型中,舒尼替尼联合多西紫杉醇、氟尿嘧啶或阿霉素等化疗药物用于乳腺癌的移植瘤动物模型,或舒尼替尼联合顺铂用于小细胞肺癌,均能观察到相加或协同的治疗作用,而且受试动物的耐受性良好,死亡率小,体重无减轻。对这种联合用药作用机制的研究表明,主要是通过抑制存在于肿瘤、间质和内皮细胞内代偿性激活的促存活信号通路,从而诱导肿瘤细胞死亡而实现的。
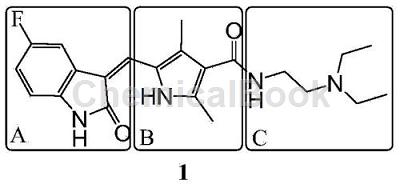
制备[2]
根据其骨架构建方式将其合成方法分为三大类,即“A+(B+C)”合成法、“B+A+C”合成法和“(A+B)+C”合成法
现有的舒尼替尼合成方法的比较
1)“A+(B+C)”合成法
目前文献报道的方法大多属于这种类型。分析其骨架的连接与吲哚环的形成先后,又可将其分为两大类。其一为先将整体骨架构建起来再形成吲哚环,其二为先将吡咯环与胺进行缩合、将吲哚环构建好,然后再使两者进行连接。
上述的方法一,例如以4-氟-2-碘苯胺为原料,经氯乙酰化、膦酰化、Horner-Emmons-Wittig反应得N-[2-(二乙胺基)乙基]-5-[(E)-2-(4-氟-2-碘代-苯胺基甲酰基)-乙烯基]-2,4-二甲基-1H-吡咯-3-甲酰胺;然后在醋酸钯和三乙胺的作用下,通过分子内5-exo型环合反应得舒尼替尼。该条合成路线较简单、但原料不易得、需回收钯催化剂、乙腈毒性较大等缺点。
上述的方法二,例如以乙酰乙酸乙酯经肟化、还原一锅法(Knorr反应)制得中间体,再水解脱羧、甲酰化、水解、与N,N-二乙基乙二胺缩合得N-(2-二乙胺基乙基)-2,4-二甲基-5-甲酰基-1H-吡咯-3-甲酰胺。另外,以对氟苯胺、水合氯醛、盐酸羟胺为原料,先酰化和肟化得到中间体,然后在浓硫酸中环合、再经黄鸣龙还原得到5-氟-1,3-二氢吲哚-2-酮。最后在三乙胺作用下利用5-氟-1,3-二氢吲哚-2-酮的3-位活泼亚甲基与N-(2-二乙胺基乙基)-2,4-二甲基-5-甲酰基-1H-吡咯-3-甲酰胺的5-位甲酰基缩合得终产物舒尼替尼。该条合成路线存在步骤较多,有些步骤的产率较低、后处理麻烦等缺点,有必要进一步改进。
2)重要中间体5-氟-1,3-二氢吲哚-2-酮(A部分)的合成方法的比较与评价
A和B以及(B+C)都是舒尼替尼的重要中间体,5-氟-1,3-二氢吲哚-2-酮的合成方法较多,列举以下两种:
方法一,用对氟苯胺、水合氯醛、盐酸羟胺为原料,先酰化和肟化,而后在浓硫酸中环合得5-氟靛红,再与水合肼反应,最后与乙醇钠反应得5-氟-1,3-二氢吲哚-2-酮。本方法步骤较多,且金属钠很活泼,制备乙醇钠时有可能会出现危险。
方法二,利用5-氟吲哚与吡啶、溴素及溴化氢形成的三溴化吡啶鎓反应得5-氟-3,3-二溴-2-吲哚酮,再经钯催化氢化脱溴得5-氟-1,3-二氢吲哚-2-酮。虽然步骤少,但原料不易得。
方法三,5-氟-2-甲基苯胺经(Boc)2O保护氨基得N-(5-氟-2-甲基苯基)氨甲酸叔丁酯,再经异丁基锂作用,通入CO2得5-氟-2-(叔丁氧甲酰胺基)苯乙酸,然后在酸性条件下脱氨基保护基并环合得5-氟-1,3-二氢吲哚-2-酮。该方法步骤较长,需进行氨基的保护,反应操作较麻烦。
主要参考资料
[1] 叶定伟, & 施国海. (2012). 中国应用舒尼替尼治疗晚期肾癌的Ⅳ期临床结果. 中华泌尿外科杂志, 33(4).
[2] 郭婕, 罗鹃, & 朱珠. (2007). 抗肿瘤新药——舒尼替尼. 中国药学杂志, 42(13), 1037-1038.
[3] 李学松, 宋毅, 龚侃, 张骞, 虞巍, & 宋刚等. (2010). 舒尼替尼治疗转移性肾脏透明细胞癌的临床研究. 中华外科杂志, 48(5), 375-377.