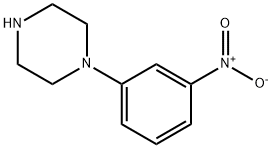
1-(3-Nitrophenyl)piperazine synthesis
- Product Name:1-(3-Nitrophenyl)piperazine
- CAS Number:54054-85-2
- Molecular formula:C10H13N3O2
- Molecular Weight:207.23
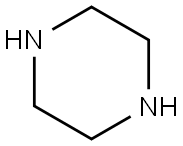
110-85-0
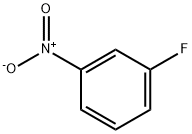
402-67-5
54054-85-2
1. 28.4 g (0.33 mol, 5.5 equiv) of piperazine is dissolved in 50 mL of DMSO. 2. 6.4 mL (60 mmol) of m-fluoronitrobenzene is added to this solution. 2. 6.4 mL (60 mmol) of m-fluoronitrobenzene was added to the above solution. 3. The reaction mixture was heated at 100°C for 60 hours. 4. 4. When the reaction is complete, cool to room temperature and slowly pour the mixture into 530 mL of water. 5. The precipitate formed was collected by filtration and the filtrate was extracted with ether (Et2O). 6. 6. The organic phases were combined, dried over anhydrous magnesium sulfate (MgSO4), and concentrated under reduced pressure. 7. The crude product was purified by column chromatography using a solvent mixture of dichloromethane (CH2Cl2) and methanol (MeOH) (9:1, v/v/v) as eluent. 8. The target component was collected and concentrated under reduced pressure to give 8.04 g of orange oil, which was crystallized as 1-(3-nitrophenyl)piperazine after standing at room temperature in 65% yield. Compound characterization data. Molecular formula: C10H13N3O2 Molecular weight: 207.26 g/mol 1H NMR (CDCl3) δ (ppm): 7.70 (t, J = 2 Hz, 1H, H-2'); 7.64 (ddd, J = 8 Hz, 2 Hz, 0.6 Hz, 1H, H-4'); 7.36 (t, J = 8 Hz, 1H, H-5'); 7.17 (ddd, J = 8 Hz, 2 Hz, 0.6 Hz, 1H, H-6') ; 3.30-3.18 (m, 4H, H-3, H-5); 3.09-2.97 (m, 4H, H-2, H-6).
110-85-0
590 suppliers
$5.00/5 g
402-67-5
273 suppliers
$6.00/5g
54054-85-2
88 suppliers
$17.67/1gm:
Yield: 72%
Reaction Conditions:
with potassium carbonate in dimethyl sulfoxide at 100; for 24 h;
Steps:
2-1; 3.1
A mixture of piperazine (12 g, 138 mmol), 1-fluoro-3-nitrobenzene (4 ml, 34 mmol) and potassium carbonate (9.60 g, 69 mmol) in DMSO (40 mL) is heated at 100°C for 24 h. The reaction is quenched with H2O (60 mL) and extracted with Et2O (3 x 20 mL). The organic layer is washed with brine (20 mL) and dried over Na2SO4 and concentrated in vacuo. The residue is chromatographied on silica gel eluting with CH2Cl2/ MeOH (94 / 6) to yield the title compound (5.15 g, 72%) as an orange solid. This solid was dissolved in CH2Cl2 and Boc2O (6 g, 130 mmol) was added. The mixture was stirred overnight. The solvent was evaporated and the residual solid was crystallized in heptane (6.80 g, 93%, mp 86°C). 1H NMR (200 MHz, CDCl3) δ 1.49 (s, 9 H), 3.22-3.27 (m, 4 H), 3.58-3.63 (m, 4 H), 7.16-7.71 (m, 4 H).
References:
INSTITUT NATIONAL DE LA SANTE ET DE LA RECHERCHE MEDICALE (INSERM) EP1683790, 2006, A1 Location in patent:Page/Page column 7; 12
206879-94-9
13 suppliers
$496.64/5MG
54054-85-2
88 suppliers
$17.67/1gm:
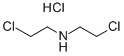
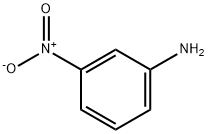
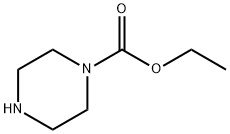
120-43-4
283 suppliers
$6.00/5g
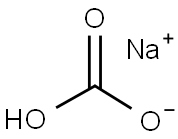
144-55-8
1410 suppliers
$10.00/25g
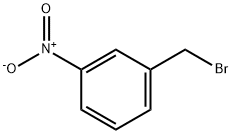
3958-57-4
252 suppliers
$17.00/10g
![Ethanamine, N-[(3,5-dichlorophenyl)methylene]-2,2-diethoxy-](/CAS/20210305/GIF/1000210-73-0.gif)
1000210-73-0
0 suppliers
inquiry
54054-85-2
88 suppliers
$17.67/1gm:
110-85-0
590 suppliers
$5.00/5 g
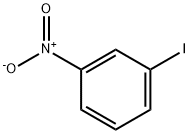
645-00-1
15 suppliers
$45.39/25g
54054-85-2
88 suppliers
$17.67/1gm: