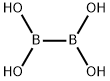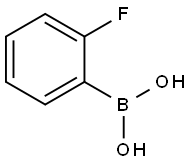
2-Fluorophenylboronic acid synthesis
- Product Name:2-Fluorophenylboronic acid
- CAS Number:1993-03-9
- Molecular formula:C6H6BFO2
- Molecular Weight:139.92
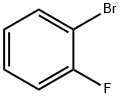
1072-85-1
1993-03-9
The general procedure for the synthesis of 2-fluorophenylboronic acid from o-bromofluorobenzene was as follows: 10.0 g (57.1 mmol) of 1-bromo-2-fluorobenzene and anhydrous tetrahydrofuran were added to a reaction flask and the mixture was subsequently cooled to -70 °C. At -70 °C, 44.2 ml (68.5 mmol) of 1.55 M n-butyllithium solution was slowly added dropwise to the reaction mixture. The temperature was maintained at -70 °C and stirring of the reaction mixture was continued for 1.5 hours. Subsequently, 21.5 g (114.2 mmol) of triisopropyl borate was added dropwise and the reaction temperature was gradually raised to room temperature and stirring was continued for 4 hours. Upon completion of the reaction, aqueous hydrochloric acid was added to the reaction mixture and extracted with dichloromethane. The organic phases were combined and the solvent was removed by distillation under reduced pressure to give the crude product of 2-fluorophenylboronic acid. The crude product was purified by column chromatography (eluent: hexane/acetone) to give 4.8 g (yield: 61%) of pure 2-fluorophenylboronic acid.
1072-85-1
467 suppliers
$5.00/10g
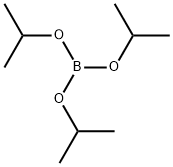
5419-55-6
388 suppliers
$12.00/5g
1993-03-9
354 suppliers
$8.00/1g
Yield:1993-03-9 85%
Reaction Conditions:
Stage #1: o-fluorobromobenzenewith n-butyllithium in tetrahydrofuran at -70; for 0.5 h;Inert atmosphere;Autoclave;Large scale;
Stage #2: Triisopropyl borate in tetrahydrofuran at -70;Large scale;
Steps:
1 Example 1 Preparation of Compound 2
Under nitrogen atmosphere, 14 kg (22.9 mol) of the compound and 20 L of THF were sequentially added to the 50 L autoclave, and the temperature was lowered to -70 ° C.Temperature control -70 the following dropping n-BuLi6.8kg, after dropping at -70 below stirring 0.5 hours. Temperature -70 below the dropBoric acid isopropyl ester 4.75kg, drop back after the natural warm night. Poured into 20L (1N) dilute hydrochloric acid, liquid, water phase and thenEA10L. The combined organic phases were washed once with saturated brine 10 L, dried over anhydrous sodium sulfate, filtered and concentratedTo give 22.4 kg of the compound in 85% yield.
References:
CN105801553,2016,A Location in patent:Paragraph 0093; 0094; 0095
1072-85-1
467 suppliers
$5.00/10g
1993-03-9
354 suppliers
$8.00/1g
1072-85-1
467 suppliers
$5.00/10g
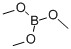
121-43-7
382 suppliers
$14.00/25mL
1993-03-9
354 suppliers
$8.00/1g
1072-85-1
467 suppliers
$5.00/10g
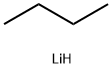
29786-93-4
0 suppliers
inquiry
5419-55-6
388 suppliers
$12.00/5g
1993-03-9
354 suppliers
$8.00/1g