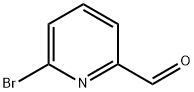
6-Bromopyridine-2-carbaldehyde synthesis
- Product Name:6-Bromopyridine-2-carbaldehyde
- CAS Number:34160-40-2
- Molecular formula:C6H4BrNO
- Molecular Weight:186.01
626-05-1
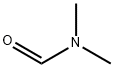
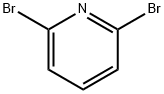
68-12-2
34160-40-2
To a 12 L three-necked flask equipped with a mechanical stirrer, addition funnel, nitrogen inlet and thermocouple sleeve was added n-butyllithium (BuLi, 619 mL, 1.55 mol). Anhydrous toluene (750 mL) was then added and the reaction system was cooled to -15 °C in an ice/methanol bath. Butylmagnesium chloride (BuMgCl, 387 mL, 0.774 mol) was added slowly over a period of 30 to 45 minutes, with the rate of addition being controlled so that the reaction temperature did not exceed 0°C. A fine white to gray suspension was formed during the reaction. The suspension was continued to be stirred at -15 °C for 30 min. During this time, 2,6-dibromopyridine (500 g, 2.11 mol) was dissolved in anhydrous toluene (3 L), which was slightly heated if necessary to promote dissolution. After stirring is complete, the 2,6-dibromopyridine solution is transferred to a dosing funnel and slowly added dropwise to the reaction flask at a rate not exceeding -5 °C (about 1.5 h). After the dropwise addition was completed, stirring of the reaction mixture was continued for 45 minutes. A small amount of the reaction solution was quenched in 20% aqueous citric acid and the extent of metal exchange was analyzed by 1H NMR and TLC (25% ethyl acetate/heptane). Toluene (750 mL) and N,N-dimethylformamide (DMF, 250 mL) were added to another 12 L three-necked flask equipped with a mechanical stirrer, nitrogen inlet, and thermocouple cannula and cooled to -15 °C in an ice/methanol bath. The initial reaction solution was transferred to the toluene/DMF mixture through a cannula at a rate not exceeding 5°C. After the transfer was complete, the reaction mixture was stirred for 45 minutes and the completion of the reaction was confirmed by TLC. The reaction solution was transferred to a partition funnel containing 4 L of water and citric acid (1 kg), stirred for 15 min and left to stratify. The organic layer was washed sequentially with 4 L of water and 4 L of saturated sodium chloride solution, dried over anhydrous sodium sulfate, and concentrated under reduced pressure to remove the solvent to afford the intermediate 6-bromopyridine-2-carbaldehyde as an off-white to yellow solid (355.7 g, 90% yield).
5315-25-3
465 suppliers
$6.00/5g
34160-40-2
343 suppliers
$6.00/1g
Yield:34160-40-2 62.3 g
Reaction Conditions:
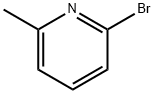
Stage #1:2-bromo-6-methylpyridine with bromine in dichloromethane;water at 10 - 50; for 10 h;
Stage #2: with hexamethylenetetramine in ethanol at 40; for 12 h;
Stage #3: with sulfuric acid;acetic acid in ethanol at 90; for 1 h;
Steps:
1.1; 1.2; 2.1; 2.2; 3.1; 3.2 (1) Preparation of a mixture of compound 2-bromo-6-bromomethylpyridine and 2-bromo-6-(dibromomethyl)pyridine
A 1-L reaction flask was charged with 2-bromo-6-methylpyridine (86 g, 0.5 mol). , 1.0 eq) and dichloromethane (172 mL, 2P), water (172 mL, 2P),The ice water bath was cooled to 10 ° C, stirred rapidly, and liquid bromine (240 g, 1.5 mol, 3.0 eq) was added dropwise, and the temperature was controlled at 10-20 ° C.After the completion of the dropwise addition, the mixture was heated to 50 ° C for 10 hours.Thereafter, it was cooled to room temperature, and the pH was adjusted to 7-8 by adding an aqueous sodium hydrogencarbonate solution.The aqueous phase was separated and extracted with methylene chloride (172 mL, 2P).The organic phase was combined and washed with saturated brine (172 mL, 2P).Drying with anhydrous sodium sulfate, suction filtration and concentration to give a mixture of 110.8 g of 2-bromo-6-bromomethylpyridine and 2-bromo-6-(dibromomethyl)pyridine.The ratio of 2-bromo-6-bromomethylpyridine and 2-bromo-6-(dibromomethyl)pyridine was determined in the liquid phase to be 6:1.(2) Preparation of compound 2-bromo-6-aldehyde pyridinea mixture of 2-bromo-6-bromomethylpyridine and 2-bromo-6-(dibromomethyl)pyridine (110.8 g, theoretical molar amount)0.5 mol, 1 eq), urotropine (140.2 g, 1.0 mol, 2.0 eq) was added to ethanol (435 mL, 4P) and heated to 40 ° C.12 hours.Thereafter, it was cooled to room temperature with acetic acid (220 mL, 2P), concentrated sulfuric acid (15 mL, 0.137 P), and then heated to 90 ° C.After an hour, it was filtered while cooling to 5 ° C, and the filter cake was washed with isopropyl ether to give 75.3 g of a yellow solid. The solid plus ethanol (75.3 mL) / water(8.37mL) recrystallization, to obtain 62.3g white solid, melting point 78.9-80.2 ° C, liquid phase 99.0%, two-step total yield67%.
References:
Anhui Haofan Biological Co., Ltd.;Wang Guichun;Lv Minjie;Liu Min CN109879815, 2019, A Location in patent:Paragraph 0058-0086
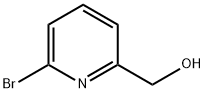
33674-96-3
258 suppliers
$8.00/250mg
34160-40-2
343 suppliers
$6.00/1g
626-05-1
506 suppliers
$9.00/10g
34160-40-2
343 suppliers
$6.00/1g
626-05-1
506 suppliers
$9.00/10g
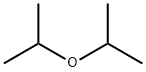
108-20-3
589 suppliers
$25.00/25mL
34160-40-2
343 suppliers
$6.00/1g
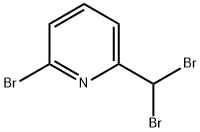
82315-66-0
11 suppliers
inquiry
34160-40-2
343 suppliers
$6.00/1g