| Identification | More | [Name]
(6R-(6alpha,7))-((Amino-1,4-cyclohexadien-1-ylacetyl)amino)-3-methyl-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid | [CAS]
38821-53-3 | [Synonyms]
(6r-(6alpha,7))-((amino-1,4-cyclohexadien-1-ylacetyl)amino)-3-methyl-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid
CEFRADIN
CEFRADINE
CEPHRADINE
CEPHRADINE HYDRATE
(6r-(6-alpha,7-beta(r*)))-ien-1-ylacetyl)amino)-3-methyl-8-oxo
(6r-6alpha,7beta(r*)))-ien-1-ylacetyl)amino)-3-methyl-8-ox
5-thia-1-azabicyclo(4.2.0)oct-2-ene-2-carboxylicacid,7-((amino-1,4-cyclohexad
7-[d-2-amino-2-(1,4-cyclohexadien-1-yl)acetamido]-3-methyl-8-oxo-5-thia-1-azab
7-[d-2-amino-2-(1,4-cyclohexadienyl)acetamide]desacetoxycephalosporanicacid
cephradin
eskacef
icyclo[4.2.0]-oct-2-ene-2-caboxylicacid
sefril
sq11436
velosef
Anspor
CEPHRADINE COMPACTED
CEPHRADINE MICRONISED POWDER
CEPHRADINE APPROX. 90% (HPLC) | [EINECS(EC#)]
254-137-8 | [Molecular Formula]
C16H21N3O5S | [MDL Number]
MFCD01743037 | [Molecular Weight]
367.42 | [MOL File]
38821-53-3.mol |
| Chemical Properties | Back Directory | [Appearance]
Solid | [Melting point ]
140-142 C | [Boiling point ]
898℃ | [density ]
1.2794 (rough estimate) | [refractive index ]
1.6320 (estimate) | [Fp ]
>110°(230°F) | [storage temp. ]
Store at 0-5°C | [solubility ]
1 M NH4OH: soluble50mg/mL | [form ]
powder | [pka]
2.63, 7.27(at 25℃) | [color ]
White | [Water Solubility ]
Soluble in water | [Stability:]
Light Sensitive | [CAS DataBase Reference]
38821-53-3(CAS DataBase Reference) |
| Safety Data | Back Directory | [Hazard Codes ]
Xi,Xn | [Risk Statements ]
R36/37/38:Irritating to eyes, respiratory system and skin .
R42/43:May cause sensitization by inhalation and skin contact .
R20/21/22:Harmful by inhalation, in contact with skin and if swallowed . | [Safety Statements ]
S26:In case of contact with eyes, rinse immediately with plenty of water and seek medical advice .
S36:Wear suitable protective clothing . | [WGK Germany ]
1 | [RTECS ]
XI0336000 | [HS Code ]
29419054 | [Hazardous Substances Data]
38821-53-3(Hazardous Substances Data) |
| Questions And Answer | Back Directory | [Overview]
Cefradine (also known as cephradine), 7-[D-2-amino-2(1,4cyclohexadien1-yl) acetamido]-3-methyl-8-0x0-5thia-l-azabicyclo[4.2.0] oct-2-ene-2-carboxylic acid monohydrate (111 is a semi-synthetic cephalosporin antibiotic. used orally, intramuscularly, and intravenously. The structure of cephradine is similar to that of cephalexin, the only difference being in the six-membered ring. Cephalexin has three double bonds forming an aromatic system while cephradine has two double bonds in the same ring. The antibacterial activity of cephradine is similar to that of cephalexin[1].

Figure1 the chemical structure of cefradine;
Cephradine is a white crystalline powder with a molecular weight of 349.4[2]. The synthesis of cephradine has been discussed[3]. Cephradine is freely soluble in aqueous solvents. It is a zwitterion, containing both an alkaline amino group and an acidic carboxyl group. In the pH range of 3-7, cephradine exists as an internal salt[4]. Cephradine is stable for 24 hr at 25" within the pH range of 2-8. Since it is stable in acidic media, there is little loss of activity in the gastric fluid; losses of less than 7% have been reported[5].
Cephradine is weakly bound to human serum proteins. The drug was less than 20% bound to the serum proteins[4]. At a serum concentration of 10-12 pg/ml, 6% of the total drug was in the protein-bound complex. Another study[6] found that at a total concentration of 10 pg/ml, 28% of the drug was in the protein-bound state; at a total concentration of 100 pg/ml, 30% of the drug was in the protein-bound state. This study also showed that the addition of serum to cephradine decreased antibiotic activity. Another study[2] showed that the protein binding of cephradine varied from 8 to 20%, depending on the concentration of the drug. However, a study by Gadebusch et al.[5] found no change in the MIC of cephradine toward either Staphylococcus aureus or Escherichia coli after the addition of human serum. | [Pharmacokinetic]
Little work has been performed on the pharmacokinetics of cephradine in subjects with normal and impaired kidney function. Further study is necessary to elucidate these parameters. A plot of cephradine serum concentration versus time indicates a bi-exponential decay. Therefore, fitting of the data for cephradine should be performed using a two-compartment open model. The effects of using a one-compartment open model analysis when a two-compartment open model is indicated were discussed previously. When the data for intravenous cephradine were fit to a two-compartment open model, the half-life was approximately 45 min. The volume of distribution was 22 liters, compared to 21-liters/1.73 m2 reported previously[13]. A value of 17 f 3.9 lited1.73 m2 also was reported[2].
The pharmacokinetics of cephradine probably will be similar to those found for cephalexin due to the similarity of their structures. The protein binding of these two compounds is not significantly different, so the volume of distributions should be similar, as was found in the reanalysis of literature data.
| [Pharmacodynamics]
Absorption
Cephradine has been administered orally, intramuscularly, and intravenously and is well absorbed using these routes. This large degree of absorption is evidenced by recoveries of approximately 100% of the administered dose in the urine[4, 7-10]. In one study[4], tritiated cephradine was administered orally to human subjects in the form of a 250-mg capsule. Urine and feces were collected and assayed for the drug; 92% of the drug was present in the urine at the end of 24 hr, while minimal amounts were present in the feces at the end of 72 hr. These findings are evidence for the claim that cephradine is well absorbed orally and apparently not excreted in the bile.
After oral administration of cephradine, the time of peak serum concentration was at 1 hr with peak concentration in the serum ranging from 6 to 7 pg/ml after a 250-mg capsule[4, 9, 10]. After the administration of 500 mg of cephradine in the form of two 250-mg capsules, peak serum levels were in the range of 11-18 pg/ml[1, 4, 7-9]. After the administration of 500 mg of cephradine as an oral suspension, a peak serum concentration of 19.5 pg/ml was found at 0.5 hr[9]. When human subjects were given 1 g of cephradine, the peak levels ranged from 15 to 24 pg/ml[1, 9].
After the administration of 1 g im, a peak level of 10.4 pg/ml at 2 hr was attained[4]. While the peak concentration of cephradine after intramuscular injection was lower than that after oral administration, the areas under the curve were identical. One advantage of the intramuscular route over the oral route was that the concentration of the antibiotic in the serum was below 1 pg/ml at 4 hr after oral administration while the concentration after intramuscular administration was 6.8 pg/ml[4].
One study[7] found that the rate of absorption after oral administration was influenced by the presence of food but that the extent of absorption was not affected. Serum levels at 30 min after administration were 7.9 pg/ml for non-fasting volunteers and 15.8 pg/ml for fasted volunteers. However, peak levels were not significantly different; values of 19.2 and 18.3 pg/ml were found for non-fasting and fasted volunteers, respectively. After intravenous administration of cephradine, the peak levels are reached immediately. In one study, a serum concentration of 56 pg/ml at 7 min after injection was attained after administration of 1 g[11]. In another study[4], the administration of 1 and 0.5 g of cephradine intravenously gave levels of 86.3 and 46.0 pg/ml, respectively, 5 min after injection. When cephradine was given as an intravenous constant infusion (0.166 g/hr), a steady-state level of 4.8 pg/ml was attained after 3 hr[11].
Distribution
In a study performed with mice[12], cephradine was widely distributed throughout the body. In this study, 50 mg/kg of tritiated cephradine was administered orally to mice and tissue levels were determined as a function of time. The levels in the stomach, small intestine, and kidneys were all above 100 pg/g of tissue. The liver had a level of more than 50 pg/g of tissue at 1 hr after administration. Almost all other body tissues had levels above l pg/g of tissue. The levels found in the brain were 0.8-2.2 pg/g of tissue in the 24-hr period of the study.
The tissue levels of cephradine in humans after oral dosing were examined[8]. The level in lung tissue 6 hr after administration of 500 mg PO was 0.46 pg/g of tissue (serum level was 0.58 pg/ml). Three hours after dosing, adipose tissue levels of 0.46-0.56 pg/g of tissue were found (serum level was 0.31-0.60 pg/ml). Cephradine levels for various other tissues were also given[8]. The volume of distribution of cephradine was reported to be 21 liter/1.73 m2[13].
Metabolism and excretion
A study in humans could not find any metabolites of cephradine[4].
When 500 mg of cephradine was administered orally to subjects having cholecystolithiasis[8], the levels of cephradine in the bile ranged from 2.2 to 41.0 pg/ml at 3-7.5 hr after administration. The major route for the elimination of cephradine is renal excretion. Cephradine is removed from the body by the processes of glomerular filtration and tubular secretion[4]. A study in which probenecid was co-administered with cephradine resulted in a prolonged half-life and elevated serum levels. The recovery of unchanged cephradine in the urine ranged from 78.3 to 95.9%[4, 7-9]. The urinary concentration of cephradine in the first 2 hr after administration of 500 mg was between 1.1 and 3.2 mg/ml[4, 7-9].
The half-lives of elimination of cephradine were 32 min[11] after intravenous administration, 40-50 min after intramuscular administration[4], and 42 min after oral administration[9]. These studies were performed in patients having normal kidney function. During a constant infusion, patients having a creatinine clearance of 125 ml/min had serum and renal clearances of cephradine of 435 and 367 ml/ min, respectively[13].
| [Indication]
Cephradine is active in vitro against a broad spectrum of gram-positive and gram-negative bacteria, including pathogenic organisms isolated in the clinic; the compound has been shown to be acid stable, and the addition of human serum had only a slight effect on the minimal inhibitory concentration (MIC) for the sensitive organisms. When given orally or subcutaneously to animals infected experimentally with a variety of pathogenic bacteria, cephradine offered effective protection[16]. In the treatment of acute infective diseases, satisfactory clinical responses to cephradine therapy have been reported by a number of investigators[14, 15, 17-19].
| [Mode of action]
Cefradine is a first generation cephalosporin antibiotic with a spectrum of activity similar to cefalexin. Cefradine, like the penicillins, is a beta-lactam antibiotic. By binding to specific penicillin-binding proteins (PBPs) located inside the bacterial cell wall, it inhibits the third and last stage of bacterial cell wall synthesis. Bacterial cell wall autolytic enzymes such as autolysins further mediate cell lysis; it is possible that cefradine interferes with an autolysin inhibitor.
| [References]
- J. F. Scholand, G. R. Hodges, R. J. Fass, and S. Saslaw, Amer. J. Med. Sci., 267, lll (1974).
- “Anspor Product Information,” AN-L2, Smith Kline and French Laboratories, Philadelphia, Pa., 1974.
- J. E. Dolfini, H. E. Applegate, G. Bach, H. Basch, J. Bernstein, J. Schwartz, and F. L. Weisinborn, J. Med. Chem., 14. 117(1971).
- E. S. Neiss, J. Ir. Med. Ass. 66,1(1973).
- H. H. Gadebusch, G. J. Miraglia, H. I. Basch, C. Goodwin, S. Pan, and K. Renz, “Advances in Antimicrobial and Antineoplastic Chemotherapy,” Vol. 1, Proceedings of the VIIth International Congress of Chemotherapy-1971, Prague, Czechoslovakia, 1972, p. 1059.
- G. Renzini, G. Ravagnan, and B. Oliva, Quad. Antibiot., 1972,l.
- C. Harvengt, P. DeSchepper, F. Lamy, and J. Hansen, J. Clin. Pharmacol., 13,36(1973).
- G. Renzini, G. Ravagnan, B. Oliva, E. Salvetti, and R. Auriti, Quad. Antibiot., 1972, 17.
- A. Zaki, E. C. Schreiber, I. Weliky, J. R. Knill, and J. A. Hubsher, J. Clin. Pharmacol., 14,118(1974).
- J. Klastersky, D. Daneau, and D. Weerts, Chemotherapy (Easel), 18.191(1973).
- C. Simon, V. Malerczyk, E. Brahnstaedt, and W. Toeller, Deut. Med. Wochenschr., 98,2448(1973).
- I. Weliky, H. H. Gadebusch, K. Kripalani, P. Arnow, and E. C. Schreiber, Antimicrob. Ag. Chemother. 5.49(1974).
- I. Weliky and A. Zoki, Eighth International Congress of Chemotherapy, Athens, Greece, 1973.
- de Mendonca, J. S., G. W. Oselka, G. C. Levi, V. A. Neto, and H. V. Lopes. 1972. Observacoes preliminares sobre an atividade terapeutica da Cefradina, nova cefalosporina, administrada por via oral. Rev. Brasil. Clin. Terap. 1:207-210.
- Estrada, F. A., B. D. Alora, and S. L. Lansang. 1972. A clinical trial of cephradine, a new cephalosporin derivative. J. Philippine Med. Ass. 48:250-254.
- Gadebusch, H., G. Miraglia, H. Basch, C. Goodwin, S. Pan, and K. Renz. 1972. Cephradine: a new orally absorbed cephalosporin antibiotic. Advan. Antimicrob. Antineoplastic Chemother. 1:1059-1062.
- Landa, L. 1972. Cephradine in the treatment of intestinal infections caused by Shigella or Salmonella organisms. Curr. Ther. Res. Clin. Exp. 14:496-502.
- Limson, B. M., R. E. Siasoco, and F. P. Dial. 1972. A new cephalosporin derivative, cephradine, in the treatment of acute infective diseases. Curr. Ther. Res. Clin. Exp. 14:101-106.
- Mitelman, A. 1972. Cefradina: una nueva cefalosporina de sintesis. Evaluacion clinicobacteriologica. Dia Med. 44:152-153.
|
| Hazard Information | Back Directory | [Description]
Cefradine is an orally bioavailable β-lactam cephalosporin antibiotic.1 It is active against S. pyogenes, E. coli, K. pneumoniae, and E. cloacae (MICs = 0.04, 9.4, 9.4, and 6.3 μg/ml, respectively). Cefradine is also active against clinical isolates of S. aureus (MICs = 0.8-6 μg/ml) and S. pyogenes (MICs = 0.1-0.4 μg/ml), as well as H. influenzae, E. coli, and K. pneumoniae (MICs = 6.2-12.5 μg/ml).2 It increases survival in mouse models of systemic lethal infection by S. pyogenes, E. coli, K. pneumoniae, or E. cloacae (ED50s = 5, 37, 122, and 50 mg/kg, respectively), as well as by penicillin-susceptible or -resistant strains of S. aureus (ED50s = 18 and 91 mg/kg, respectively).1 Formulations containing cefradine have previously been used in the treatment of respiratory and urinary tract infections, skin infections, and otitis media. | [Description]
In cephradine, an interesting drug design device has been used. The aromatic ring in the ampicillin side
chain has been partially hydrogenated by a Birch reduction such that the resulting molecule is still planar
and π-electron excessive but has no conjugated olefinic linkages. It is comparatively acid stable and,
therefore, is rapidly and nearly completely absorbed from the GI tract. Cephradine has the useful characteristic that it can be used both orally and IM so that parenteral therapy can be started in an
institutional setting and then the patient can be sent home with the oral form, thus avoiding the risk of
having to establish a different antibiotic. This is consistent with the present economics requiring sending
patients home earlier than some physicians prefer. Unfortunately, however, for other reasons the IM and
intravenous (IV) versions of cephradine are no longer available in the United States. | [Description]
This drug is very similar to cephalexin in its antimicrobial activity and
in most other respects (Moellering and Swartz, 1976). Unlike
cephalexin, for which a parenteral preparation is not generally available, cephradine is marketed in some countries (for example,
Portugal) for both oral and parenteral use. | [Chemical Properties]
Solid | [Originator]
Sefril,Squibb,Switz. | [Uses]
A semi-synthetic cephalosporin antibiotic that is used to study the effect of expression, binding, and inhibition of PBP3 and other penicillin-binding proteins (PBPs) on bacterial cell wall mucopeptide synthesis | [Uses]
Cephalosporin antibacterial. | [Uses]
Cephradine is a first generation cephalosporin antibiotic. Cephradine has broad spectrum of bactericidal activity against infections caused by Streptococcus, Staphylococcus, Diplococcus pneumoniae, Es
cherichia, Klebsiella, Salmonella, and indole-negative Proteus. | [Application]
Cephradine was synthesized by the Squibb Institute of Medical Research in 1971. It shows almost the same antibacterial activity and pharmacokinetic properties as cephalexin. Cephradine has been used for therapy of urinary and respiratory tract infections caused by Staphylococcus, Streptococcus, Escherichia coli, Klebsiella, and Proteus mirabilis. | [Definition]
ChEBI: A cephalosporin with a methyl substituent at position 3, and a (2R)-2-amino-2-cyclohexa-1,4-dien-1-ylacetamido substituent at position 7, of the cephem skeleton. | [Manufacturing Process]
In a first step, D-2-amino-2-(1,4-cyclohexadienyl)acetic acid is obtained as
follows. A solution of 11.0 g (72.7 mmol) of D-phenylglycine in 900 ml
distilled ammonia (which has been treated with 45 mg lithium after distillation
to destroy traces of moisture) is slowly diluted with 370 ml dry ten-butyl
alcohol.
Over a period of hours, 1.65 g lithium (3.27 eq) is added in small portions
until a permanent blue color is obtained. The blue reaction mixture is then
treated with 38 g of triethylamine hydrochloride. The ammonia is allowed to
evaporate at room temperature overnight and the residual solvent is
evaporated at reduced pressure. The white residue is taken up in a small
amount of methanol-water and added to 4 liters of cold 1:1 chloroform-acetone to precipitate the crude product. After 20 minutes stirring the
suspension is filtered and the white filter cake dried in vacuo; the filter cake is
then pulverized and submitted once more to the precipitation process from
1:1 chloroform-acetone.
The white, crystalline product, 11.8 g, MP 297°C (dec), [α]D -89.7 (2 N NaOH)
is quantitatively obtained but is slightly contaminated with lithium chloride,
0.6% ionic chlorine being found by analysis.
The product of a second step is the methyl acetoacetic ester enamine of N-2-
amino-2-(1,4-cyclohexadienyl)acetic acid sodium salt. 306 mg D-2-amino-2-
(1,4-cyclohexadienyl)acetic acid (2.00 mmol) are dissolved by warming in a
solution of 108 mg of NaOCH3 (2.00 mmol) in 4.3 ml reagent grade MeOH.
255 mg (0.24 ml, 2.20 mmol) methyl acetoacetate are added and the mixture
refluxed for 45 minutes. The MeOH is almost totally stripped off in vacuo. Five
milliliters benzene are added and distilled off to a small residual volume. The
addition and distillation of benzene is repeated to insure complete removal of
the MeOH and water. The product crystallizes out overnight from a small
residual volume of benzene. It is filtered off, washed with benzene, and dried in vacuo. Yield 463 mg.
Then 3-deacetoxy-7-aminocephalosporanic acid is condensed with the above
described sodium salt in the presence of triethylamine to give cephradine. | [Brand name]
Anspor (GlaxoSmith-
Kline); Velosef (Bristol-Myers Squibb). | [Therapeutic Function]
Antibiotic | [Antimicrobial activity]
Cephradine. A semisynthetic cephalosporin available in both
oral and injectable forms. The antibacterial spectrum and
susceptibility to β-lactamases are almost identical to those of
cefalexin .
It is almost completely absorbed when given by mouth. A
500 mg oral dose achieves a concentration of about 18–20
mg/L after 1 h. The peak is delayed and reduced by food, but
the half-life is not altered. Intramuscular administration of
1 g results a plasma concentration of 10–12 mg/L within 2 h.
The plasma half-life is around 1 h and protein binding low.
Concentrations of up to 40% of those simultaneously
found in the serum have been demonstrated in lung tissue.
Penetration into the CSF is poor. Levels in sputum were
about 20% of those simultaneously present in the plasma following
a 1 g oral dose and similar levels have been found in
bone. Breast milk concentrations approaching 1 mg/L have
been found after 500 mg orally every 6 h and similar concentrations
have been found in amniotic fluid. Cord blood concentration
is said to be similar to that in the maternal blood.
It is excreted unchanged in the urine mostly in the first 6 h,
achieving concentrations exceeding 1 g/L. Probenecid markedly
increases the plasma concentration and delays the peak.
There is some biliary excretion.
The parenteral forms may give rise to local pain or thrombophlebitis.
Other side effects common to cephalosporins
have been described. In some patients Candida vaginitis has
been troublesome.
Clinical use is similar to that of cefalexin, but it has been
largely superseded by later cephalosporins. | [Clinical Use]
Cephradine (Anspor, Velosef) is the only cephalosporinderivative available in both oral and parenteral dosageforms. It closely resembles cephalexin chemically (it maybe regarded as a partially hydrogenated derivative ofcephalexin) and has very similar antibacterial and pharmacokineticproperties.
It occurs as a crystalline hydrate that is readily soluble inwater. Cephradine is stable to acid and absorbed almostcompletely after oral administration. It is minimally proteinbound and excreted almost exclusively through the kidneys.It is recommended for the treatment of uncomplicated urinarytract and upper respiratory tract infections caused bysusceptible organisms. Cephradine is available in both oraland parenteral dosage forms. | [Synthesis]
Cefradin, [6R-[6|á,7|?(R)]]-3-methyl-8-oxo-7-[(amino-1,4-cyclohexadien-1-
ylacetyl)amino]-5-thia-1-azabicyclo[4.2.0]oct-2-en-2-carboxylic acid (32.1.2.13), is a close analog of cephalexin and differs in that the phenyl group in phenylglycine is partially hydrated to a 1,4-cyclohexadienyl moiety.
It is synthesized from phenylglycine, which is partially
reduced by lithium in liquid ammonia, which forms 1,4-cyclohexadienylglycine (32.1.2.11),
and the amino group in this compound is protected by reacting it with methyl acetoacetate in
the presence of sodium methoxide. The resulting salt (32.1.2.12) is transformed into a mixed
anhydride by a reaction with ethyl chloroformate in triethylamine, and reacted with deace�toxylated 7-aminocephalosporanic acid, which gives cefradin (32.1.2.13).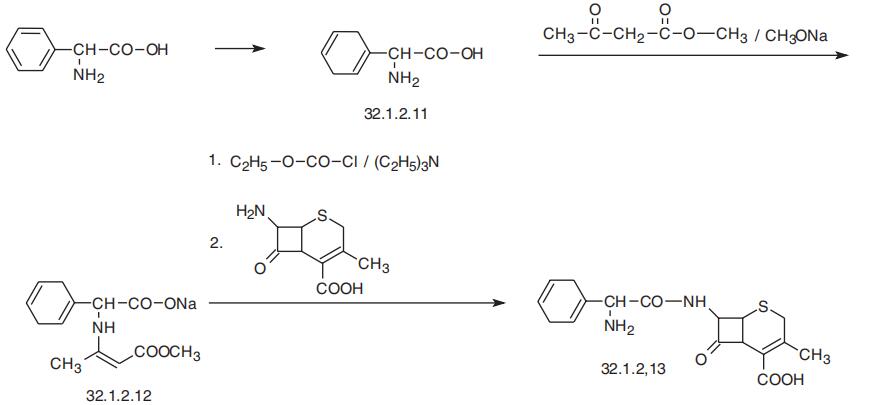
| [Drug interactions]
Potentially hazardous interactions with other drugs
Anticoagulants: effects of coumarins may be
enhanced. | [Metabolism]
Cefradine is excreted unchanged in the urine by
glomerular filtration and tubular secretion, over 90% of
an oral dose or 60-80% of an intramuscular dose being
recovered within 6 hours. Probenecid delays excretion. | [storage]
Store at -20°C |
|
|