近日,勃林格殷格翰的乙磺酸尼达尼布(Nintedanib Esylate,Ofev®)的进口上市申请审评完毕,目前处于审批阶段,有望在中国获批用于治疗特发性肺纤维化(Idiopathic pulmonary fibrosis, IPF)。
从1998年确定靶点、立项,到2014年获批上市,经历了16年的风雨,尼达尼布终于上市!其独特的作用机理让IPF患者收益良多。2016年Ofev销售额的同比增长70%,达6.78亿美元。以此增长,预计Ofev有望于2017年突破10亿美元成为IPF领域中的重磅炸弹。市场前景,异常广阔,勃林格殷格翰的研发人员功不可没。那么,研发过程中尼达尼布的靶点设计及路线选择,对我们有什么借鉴意义呢?
作用靶点
乙磺酸尼达尼布是一种有效的口服小分子酪氨酸激酶抑制剂,也称为三重血管激酶抑制剂,抑制血管生成中涉及的三种主要信号通路。乙磺酸尼达尼布靶向血管内皮细胞生长因子受体(Vascular Endothelial Growth Factor Receptor,VEGFR)家族,成纤维细胞生长因子受体(fibroblast growth factor receptor,FGFR)家族,血小板衍生生长因子受体(platelet-derived growth factor receptor,PDGFR)家族以及Src和Flt-3激酶介导的促血管生成和促纤维化途径(图1)。
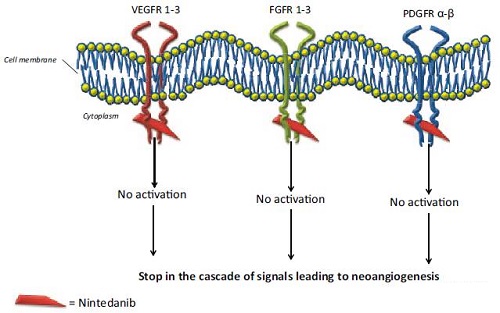
图1 尼达尼布作用靶点
结构与合成
1998年,勃林格殷格翰成立了开发用于治疗癌症的血管生成抑制剂的优化项目。起初,项目的关键目标是:选择性抑制血管内皮生长因子2(VEGFR-2)超过其他激酶、有效抑制内皮细胞增殖、良好的口服生物利用度和证明在体内肿瘤异种移植物中的活性。研究人员艰苦奋斗,寻寻觅觅,优中选优,最终筛选出BIBF1120,通用名为乙磺酸尼达尼布,一种6-甲氧基羰基取代的吲哚酮衍生物(图2)。
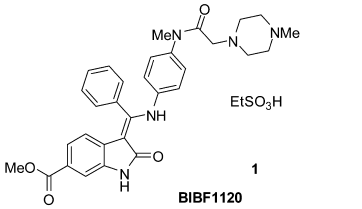
图2 乙磺酸尼达尼布
乙磺酸尼达尼布的合成涉及两个重要中间体(6)和(9),全合成路线详见图3。中间体(6)和(9)合成路线如下:
将4-氯-3-硝基苯甲酸酯(3)和丙二酸酯经烃化反应得硝基苯类化合物(4),(4)经还原、环化反应得吲哚类化合物(5),(5)经乙酰化、酯缩合、消除反应得中间体(6)。
中间体(9)是一种哌嗪类衍生物,其合成可以采用一锅法。以N-甲基-4-硝基苯胺为原料,经酰化、N-烷基化、催化氢化还原反应获得。
(6)和(9)经共轭加成-消除反应得尼达尼布游离碱2(pKa=7.9)。2在甲醇中与乙磺酸经成盐反应得乙磺酸尼达尼布(1)。(1)具有以下理化性质:mp=305℃,log P=3.0,高溶解度(水,>20 mg/mL)。
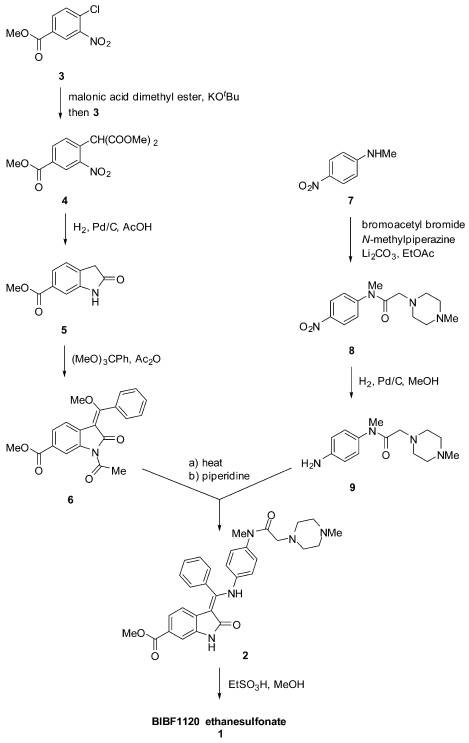
图3 乙磺酸尼达尼布合成路线
药物化学和构效关系(SAR)
最初,基于高通量筛选出了VEGFR-2抑制剂。同时,相关激酶项目(CDK4)衍生物的选择性测试中发现化合物10(见表1)的VEGFR-2抑制力不强但已具有细胞活性。并且对另一种近缘激酶CDK4,完全没有作用,也就是说有很高的选择性。当在小激酶选择性测试时,化合物10显示出明显优于其他结构的特异性模式。这种选择性的可能是因为羟吲哚母核的6位上的取代基。研究人员假设,这个位置的取代基应该占据ATP结合区域的“specificity pocket”(见图4)。后来该假设被尼达尼布的X-射线结构和VEGFR-2的激酶结构域所证实。研究人员认为化合物的选择性是首要条件,抑制活性可以进一步优选。在这种思路指导下,选择化合物10作为Lead,进一步衍生化研究。
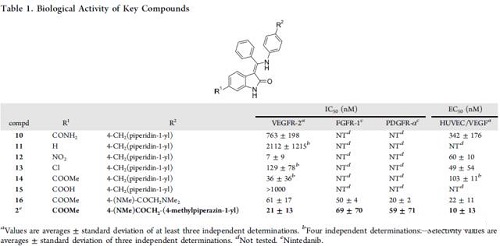
表1 关键化合物10的生物活性
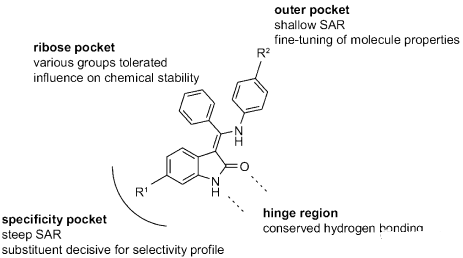
图4 6-取代氧吲哚的构-效关系
合成的场战役(synthesis campaign)是得到SAR,提高化合物10的生物活性。图4显示了总体的SAR。用较小的烷基取代核糖“pocket”中的中央芳基,不利于分子的化学稳定性,这可能是因为只有苯基部分可以采用垂直于分子其余部分的无应变构象。这种垂直构象破坏了整体平坦的化合物形状,并且可能是乙磺酸尼达尼布水溶性好的原因之一。尽管无取代基的化合物11(表1)对于相关激酶如CDK4,InsR和IGF1R(1μM)显示出较低的选择性,但是6位的取代处理可以使化合物良好的进入或退出激酶。最有效的变异是6-硝基取代(化合物12,表1),但是因为硝基的诱变性能,放弃。氯取代的化合物如13进行了进一步研究,但由于选择性较小,最终终止。
尽管酯取代化合物(如化合物14)是有效的抑制剂,酯酶的代谢降解是一个明显的风险。只有当14和衍生物在啮齿动物中表现出可接受的口服利用度时,才将其进一步优化。当对40个激酶进行选择性测试时,化合物14之类显示出优异的激酶特异性。选择性测试包括对FGFR和PDGFR的抑制作用,这被认为对化合物的整体抗血管生成作用有益。6-羧基氧吲哚(如15)对VEGFR-2,FGFR或PDGFR受体没有显示活性。
在R2处优化外缘的激酶口袋的芳环取代,能得到直接的效果。表面的SAR可以微调细胞活性和溶解性等性质。对于多数化合物,都能很好地抑制人脐静脉内皮细胞(HUVEC)增殖。R2的优化得到了一组高效的、有选择性和口服的化合物,如BIBF1000(16)。表2可以清晰的看出研究人员的优化路径。在评估异种移植实验(见靶点结合),化学制造控制表征,一般药理学和耐受性测试(见毒理学)中进行后,2001年完成了优化,乙磺酸尼达尼布(2)进入临床前研究阶段。
表2 Lead化合物10的优化与结果

靶点结合
乙磺酸尼达尼布的临床前研究采用体外抑制实验,证明其明显的、高选择性的药理活性(见表3)。靶点激酶包括所有3个VEGFR亚型,FGFR1、2和3以及PDGFR-α和β。同时靶向这三种不同的促血管生成受体可能会增强其抗肿瘤作用,并克服VEGF-和VEGFR-2靶向药物抗性。尼达尼布还抑制Flt-3和Src家族(Src,Lyn和Lck),可能具有治疗诸如血液病的潜力。
实验发现VEGFR-2转染的NIH3T3细胞暴露于尼达尼布仅1小时,VEGFR-2的体外抑制持续至少32h,显著的抑制靶点激酶。采用脐静脉(HUVEC)和皮肤微血管(HSMEC)(EC 50 <10 nM)的内皮细胞,证明乙磺酸尼达尼布抑制VEGF刺激的细胞增殖。该抑制途径是通过抑制MAPK和Akt磷酸化。
乙磺酸尼达尼布还抑制FGF诱导的HUVEC增殖,PDGF诱导的周细胞和人脐动脉血管平滑肌细胞(HUASMC)。然而,对人上皮癌细胞系的增殖没有观察到抑制作用,没有检测到VEGFR,FGFR或PDGFR的表达。
在一系列体外肿瘤异种移植模型中,乙磺酸尼达尼布有显著的抑制活性,包括NSCLC(Calu-6),人肾细胞癌(RCC)(Caki-1),结肠直肠(HT-29),卵巢(SKOV-3 )和前列腺癌(PAC-120)。
在NSCLC(H460)的异种移植物中,乙磺酸尼达尼布与多西紫杉醇或培美曲塞显示出协同效应。与多西紫杉醇的组合使肿瘤缩小至仅为对照组的27%,而相同剂量的单用尼达尼布或多西紫杉醇治疗效果差(肿瘤生长抑制率分别为75%和66%)。对Calu-6细胞尼达尼布和培美曲塞的组合有相似结果。
在一系列临床前研究中评估了乙磺酸尼达尼布抑制VEGFR,PDGFR和FGFR的抗纤维化作用。乙磺酸尼达尼布在体外抑制正常人肺成纤维细胞的PDGFR-α和-β的活性和增殖,并抑制IPF患者和对照供体的PDGF-BB-,FGF-2-和VEGF诱导的人肺成纤维细胞增殖。
在IPF的两种不同的小鼠模型中,乙磺酸尼达尼布有抗炎作用。通过组织学分析,IPF小鼠模型揭示了乙磺酸尼达尼布抗纤维化作用是显著降低肺胶原和纤维化。
表3乙磺酸尼达尼布体外抑制激酶的活性

非临床药物代谢药代动力学
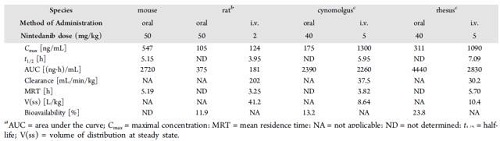
使用CD1小鼠,大鼠,猕猴和恒河猴的临床前毒性研究时,评估了乙磺酸尼达尼布的PK曲线。大鼠口服生物利用度约为12%,猴子的口服生物利用度为13%~24%(表4)。尼达尼布在大鼠中不完全吸收,并且有首过代谢。胃酸分泌的抑制对乙磺酸尼达尼布的吸收没有影响,盐酸盐或乙磺酸盐具有相似的血浆浓度与时间曲线。
乙磺酸尼达尼布经吸收后,在所有研究的物种中广泛地分布在组织中,稳定状态时V(SS)> 8 L/kg(表4)。乙磺酸尼达尼布具有浓度依赖性,血浆蛋白在50至2000 ng/mL的浓度范围内结合,白蛋白是主要结合蛋白。乙磺酸尼达尼布的结合比例,在人血浆中为97.8%,小鼠血浆97.2%,大鼠血浆98.5%。
乙磺酸尼达尼布的消除,主要是通过酯解和随后的代谢途径。同时考虑血浆和排泄物的代谢模式,尼达尼布的代谢细分成以下主要反应:
(1)将6-取代氧吲哚的甲基酯的酯解;
(2)哌嗪部分的氧化N-去甲基化;
(3)酯解和氧化N-去甲基化。
主要排泄途径为胆汁代谢产物,经口给药后排泄较少,小鼠药物相关放射性剂量为2.1%,尿液排泄量为1.2%。
表4乙磺酸尼达尼布在部分动物中的PK数据
疾病与机制
目前的抗血管生成药物的局限性越来越明显,有效性不足和耐药性限制了VEGF靶向疗法的使用。诸如高血压、出血、胃肠穿孔的风险,皮肤病学和粘膜炎等毒性也可能限制了VEGF或VEGFR抑制剂的使用。
乙磺酸尼达尼布作为三种促血管生成途径受体家族(VEGFR,FGFR和PDGFR)的有效抑制剂,应用范围更广。
特发性肺纤维化(IPF)是一种慢性和进行性纤维化肺疾病,由于缺乏有效的治疗,确诊后的中位生存数只有2-3年。IPF的特征在于成纤维细胞/肌成纤维细胞不受控制的增殖和分化,以及在肺间质和肺泡空间,引起咳嗽和呼吸困难,导致呼吸衰竭。
IPF的致病机制尚未完全明确,但是乙磺酸尼达尼布的靶点——许多酪氨酸激酶受体被作为介质。FGF-1和FGFR-2在IPF患者肺中的各种细胞表达。此外,FGF-2刺激I肺成纤维细胞增殖,并且抑制FGF信号可以降低肺纤维化,改善IPF小鼠模型的存活率。PDGF造成肌成纤维细胞的增殖,迁移和存活。VEGF表达也与哮喘患者的上皮性纤维化相关,抗VEGF治疗在IPF小鼠模型中显示出一些功能益处。
临床实验数据
除获批用于治疗IPF外,乙磺酸尼达尼布还获欧洲EMA批准用于治疗非小细胞肺癌(NSCLC)。IPF的关键3期临床研究数据见表5。
表5 乙磺酸尼达尼布的IPF关键3期临床研究数据

上市与前景
从启动新型血管新生抑制剂项目,已经超过15年。花费了大量精力来研究和评估初始化合物,乙磺酸尼达尼布代表了这一努力的成果,并且在审评阶段获得一定的优惠政策,如快速审评通道、优先审评、孤儿药、突破性治疗等。下表6为乙磺酸尼达尼布在美欧日的上市时间。
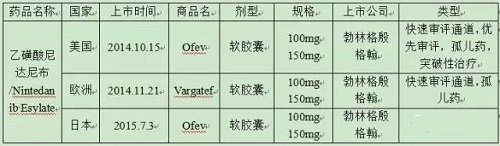
表6乙磺酸尼达尼布美欧日上市时间
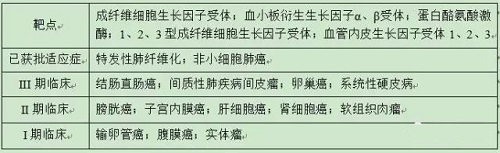
乙磺酸尼达尼布具有多个靶点(表7),体内活性高。除已获批特发性肺纤维化和非小细胞肺癌2个适应症外,尼达尼布正在开发新的适应症,市场前景广阔,见表7。
表7 尼达尼布靶点及适应症信息
参考文献
1. Nintedanib: From Discovery to the Clinic
2. 比癌症还凶险的致命性肺部疾病迎来新的竞争者,国内保卫战即将打响!
3. http://www.pharmaguider.cn/big_data_show.php?id=175