背景技术
β-突厥烯酮(β-Damascenone),又叫β-大马烯酮,化学名为4-(2,6,6-三甲基-1,3-环己二烯)-2-丁烯-4-酮,为淡黄色至黄色液体,CAS:23696-85-7。因其具有诱人的玫瑰香气,兼有覆盆子样的果香和扩散力,成为珍贵的香原料。该原料首先由Ohloff在保加利亚突厥玫瑰油中发现其微量存在,且是关键性的香气成份。近年来随着香料工业的发展、合成工艺技术改进等,其价格亦有所下调,使用量也逐年增加。β-突厥烯酮类香料不仅在配制高级香水和调配化妆品香精中用作珍贵香原料,而且已被应用到食品香精中,同时,还可在烟草中改善香烟的韵味,使烟味变得更为柔和舒适。
β-突厥烯酮的合成路线有很多,国外报道较多,常见有一定价值的合成方法如下:
1.文献Helv.Chim.Acta.1970,53(3),541报道了以柠檬醛为原料经β-环柠檬醛,丙烯溴格式加成,氧化,溴代消除得到,具体反应式如下:
反应a柠檬醛环化需要用到高毒的苯胺,会产生大量的胺氮废水,反应b烯丙基溴没有商品化,需要自制,自制工艺也很麻烦,反应c氧化需要用到二氧化锰或者含铬试剂,最后反应d溴代用到NBS,总之该工艺会产生大量的三废,工艺复杂难操作,很难工业化生产。
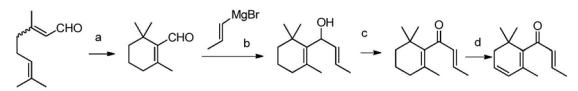
2.Snowden等人(Helv.Chim.Acta.1987,70,1858)报道了以藏红花酸甲酯为原料合成,与两分子丙烯氯格氏试剂加成,而后在KH/HMPA的条件下得到β-突厥烯酮的方法,具体反应式如下所述:

原料藏红花酸甲酯合成工艺较复杂,不易获得,反应f用到了KH/HMPA,很危险,其工业化易燃易爆的风险非常大。
3.文献(J.C.S.Perkin 1,1975,1727-1736)报道了Diels-Alder合成路线,溴代异丙叉丙酮与1,3-戊二烯环加成,再消除溴化氢,与乙醛aldol缩合脱水得到β-突厥烯酮,具体如下:

该工艺原料溴代异丙叉丙酮难得,反应j用到了N-甲基苯胺溴化镁,会产生大量的胺氮废水。
4.日本专利JP74-7556和美国专利US4250332报道了三甲基环己烯酮与甲基丙炔醇在锂氨存在下进行加成反应,制得二醇,然后将此二醇与80%甲酸回流1/2小时,得到β-突厥烯酮。

该工艺原料2,6,6-三甲基环己烯酮不易获得,氨基锂工艺操作风险较大。
5.Ohloff等人(Helv.Chim.Acta,1973,56,310-320)报道Bǜchi-vederas重排反应路线制备β-突厥烯酮,利用β-紫罗兰酮为原料,先肟化,再制成异噁唑、环氧化后再开环,最后用酸处理制得β-突厥烯酮。

该工艺反应p还原用到金属钠和液氨,安全风险较大。
鉴于以上工艺的种种弊端,还需要开发一些更适合工业化生产的β-突厥烯酮合成路线。
发明内容
本发明的目的在于克服现有技术的不足之处,提供了一种条件温和、便于操作、成本低、原料易得的合成β-突厥烯酮的方法,本工艺经过香料α-突厥酮作为中间体,可以同时合成α-突厥酮及β-突厥烯酮。
本发明解决其技术问题所采用的技术方案是:
一种合成β-突厥烯酮的方法,包括:
1)α-环香叶酸的合成:
步骤1:往反应容器中加入柠檬醛、还原剂、溶剂和磷酸二氢钠水溶液,温度10~40℃下往其中滴加亚氯酸钠溶液;滴加完毕后,室温下搅拌反应16~24h;分出有机相,水相用萃取剂萃取,合并有机相,除去其中的溶剂和萃取剂;然后加种碱中和,搅拌0.2~1h,分液,水相用萃取剂萃取,弃去有机相,萃取后的水相用种酸调节pH值至1.8~2.2,分出有机相,水相用萃取剂萃取,合并有机相,浓缩得粗品;
步骤2:往反应容器中加入步骤1得到的粗品,加入第二溶剂,加热至100~115℃,往其中滴加第二种酸,保温搅拌6~12h;往其中加入水,搅拌,分出有机相,水相用第二萃取剂萃取,合并有机相,浓缩得粗品,重结晶若干次,得α-环香叶酸;
2)α-突厥酮的合成:
步骤3:反应容器中加入步骤2得到的α-环香叶酸和第三溶剂,往其中滴加氯代试剂,回流反应2~4h,除去氯代试剂和部分第三溶剂;降至室温,保护气氛下,往其中滴加第二种碱,滴加完毕后,升温至140~160℃,回流进行消除反应5~8h;降至室温,除去析出的固体,滤液浓缩,得环香叶烯酮;
步骤4:反应容器中加入镁屑和第四溶剂;保护气氛下,将卤代烃溶于第四溶剂中,将其向反应容器中加入少许引发后,滴加剩余溶液至反应容器中,维持温度不超过42℃,滴加完毕后,室温下搅拌0.5~2h,得到格式试剂;格氏试剂降温至-5~5℃,往其中滴加步骤3得到的环香叶烯酮,控制温度不超过25℃,滴加完毕后,搅拌0.5~2h进行格氏加成;滴加淬灭剂淬灭至悬浊沙状固体,固液分离,异构化溶剂洗涤固体,滤液浓缩得粗品,往其中加入异构化溶剂和第三种酸,保护气氛下,加热回流反应10~18h,加第三种碱中和洗涤至弱碱性,分液,水相用异构化溶剂萃取,合并有机相,浓缩得α-突厥酮;
3)β-突厥烯酮的合成:
步骤5:反应容器中加入步骤4得到的α-突厥酮、盐、第五溶剂,搅拌下,冷却至8~12℃;滴加环氧化试剂,温度不超过25℃,滴加完毕后,升至常温,继续搅拌过夜进行环氧化反应;加入水,搅拌分层,水相用第三萃取剂萃取,合并有机相,用淬灭还原剂洗涤,分液,有机相浓缩,得环氧突厥酮;
步骤6:反应容器中加入第四种碱、步骤5得到的环氧突厥酮、第六溶剂,保护气氛下,加热回流5~6h,反应完全后,加水,除去第六溶剂,分液,水相用第四萃取剂萃取,合并有机相,加入干燥剂干燥,过滤,浓缩得羟基突厥酮;
步骤7:反应容器中,加入第四种酸、第七溶剂、步骤6得到的羟基突厥酮,保护气体保护下,加热至回流11~13h,反应完毕后,加入第五种碱至弱碱性,分出有机相;除去第七溶剂,浓缩得β-突厥烯酮。
合成方法
1)α-环香叶酸的合成:
步骤1:以柠檬醛为原料,经过改进过的Pinnick氧化反应得到香叶酸中间体。具体地:
往5L的三颈瓶中加入柠檬醛(100g)、双戊烯(446g)、叔丁醇(486g),加入902g的16.8%的磷酸二氢钠水溶液,15℃下往其中缓慢滴加20%亚氯酸钠溶液379.6g,温度不高于35℃;滴加完毕后,室温下搅拌反应16h;待原料反应完全,分液,分出上层有机相,下层水相用甲苯萃取(2x100mL),合并有机相,旋蒸,除去其中的叔丁醇和甲苯;然后10%的氢氧化钠溶液400g,搅拌0.5h,分液,水相用甲苯萃取(2x100mL),弃去有机相,萃取后的水相用31%的硫酸调节pH值至2.0,分出有机相,水相用甲苯萃取(2x50mL),合并有机相,旋蒸,得粗品117.8g,为香叶酸,含量77.4%(两个异构体之和),直接投入下一步反应;
步骤2:香叶酸采用85%的磷酸,加热至100℃环化,结晶得到α-环香叶酸。具体地:
往反应瓶中加入117g步骤1得到的香叶酸粗品,加入甲苯200g,加热至100℃,往其中滴加85%的磷酸20g,保温搅拌8h;往其中加入水200g,搅拌,分出有机相,水相用甲苯萃取(2x100mL),合并有机相,旋蒸浓缩得粗品106g,加入环己烷多次重结晶,得α-环香叶酸,两步收率70%;
2)α-突厥酮的合成:
步骤3:环香叶酸在氯化亚砜的条件下,制备酰氯,浓缩除去多余的氯化亚砜和甲苯,直接加入三乙胺作碱,加热消除,蒸馏得到中间体环香叶烯酮。具体地:
1L三口瓶中,加入47g步骤2得到的α-环香叶酸,甲苯300g,装好回流冷凝管,干燥管,尾气碱液吸收装置。往反应瓶中滴加氯化亚砜82.2g,回流反应2~4h,常压蒸馏蒸出残留的氯化亚砜和部分甲苯;降至室温,氮气氛下,往其中滴加三乙胺34g,滴加完毕后,升温至145~150℃,回流进行消除反应6h;降至室温,析出大量固体,抽滤除去析出的固体盐,滤液减压蒸馏,得环香叶烯酮21.4g,收率51%;
步骤4:烯丙基氯格氏试剂制备好后,往其中滴加环香叶烯酮,控制好温度,反应完毕后,饱和氯化铵淬灭反应,抽滤,浓缩,加入乙酸乙酯、催化量对甲苯磺酸异构化,浓缩蒸馏,得到α-突厥酮。具体地:
往150mL的反应瓶中加入镁屑3.4g,THF 30g;置换氮气,将烯丙基氯10.1g溶于THF50g中,将其向反应瓶中加入少许引发后,缓慢滴加剩余溶液至反应瓶中,维持温度不超过42℃,滴加完毕后,室温下搅拌1h,得到格式试剂;
格氏试剂降温至冰浴,往其中滴加步骤3得到的环香叶烯酮9g,控制温度不超过25℃,滴加完毕后,搅拌1h进行格氏加成;滴加饱和氯化铵溶液(19.3g)淬灭至悬浊沙状固体,抽滤,乙酸乙酯洗涤固体,滤液浓缩得12.9g粗品,往粗品中加入乙酸乙酯70g、对甲苯磺酸0.6g,氮气氛下,加热回流反应12h,加20%的碳酸钠溶液20g洗涤至弱碱性(pH值约8.0),分液,水相用乙酸乙酯萃取(2x15g),合并有机相,减压浓缩,蒸馏得α-突厥酮8.2g,收率71%;
3)β-突厥烯酮的合成:
步骤5:α-突厥酮在乙酸钠的二氯甲烷溶液中,滴加20%的过氧乙酸溶液,反应完毕后,用硫代硫酸钠淬灭反应,浓缩,减压蒸馏得环氧突厥酮。具体地:
往5L的三口烧瓶中,加入步骤4得到的α-突厥酮225g,乙酸钠192g,二氯甲烷1400g,搅拌下,冰浴下冷却至10℃;滴加20%过氧乙酸水溶液946g,温度不超过25℃,滴加完毕后,升至常温,继续搅拌过夜进行环氧化反应;加入水450g,搅拌分层,水相(上层)用二氯甲烷萃取2次(2x150g),合并有机相,用10%的硫代硫酸钠溶液(375g)洗涤,分液,有机相浓缩,得粗品282.8g,减压蒸馏得产品环氧突厥酮217.8g,收率89%;
步骤6:环氧突厥酮在碳酸钾/甲醇溶液中加热回流开环,反应完毕减压旋去甲醇,乙酸乙酯萃取,浓缩,得到羟基突厥酮粗品,无需纯化可直接投入下一步反应。具体地:
往2L的单口瓶中,加入72g碳酸钾、217g步骤5得到的环氧突厥酮、1085g无水甲醇,氮气氛下,加热回流5.5h,反应完全后,加水500mL,旋蒸除去甲醇,分液,水相用乙酸乙酯萃取(2x150mL),合并有机相,加入元明粉干燥,过滤,旋蒸浓缩,得羟基突厥酮粗品228g,直接投入下一步反应;
步骤7:羟基突厥酮在二氯甲烷溶剂中,对甲苯磺酸做催化剂,加热回流,反应完毕后,饱和碳酸氢钠洗涤,浓缩,蒸馏得到最终产品β-突厥烯酮。具体地:
3L的三口烧瓶中,加入41.7g对甲苯磺酸、1.6kg的二氯甲烷,228g步骤6得到的羟基突厥酮,氮气保护下,加热至回流12h,反应完毕后,加入500mL的饱和碳酸氢钠洗涤至弱碱性(pH值约8.0),分出有机相;旋蒸除去二氯甲烷,得粗品218g,减压蒸馏,得β-突厥烯酮126.8g,两步收率64%。