血管紧张素2高的原因
【血管紧张素2】
血管紧张素2(AngII)是肾素血管紧张素系统(RAS)中最主要的生物活性物质,AngII收缩血管的作用是去甲肾上腺素的40倍,是己知天然存在的升压物质中最强者之一。血管紧张素II可与所有组织中的AngII受体结合。作为系统的重要效应肽,主要通过与血管紧张
素1型(AT1)受体和血管紧张素型2(AT2)受体的结合发挥其生物活性效应。
AT1受体:能被Losartan和DDT阻断的受体为AT1受体,AT1受体又分为AT1A受体和AT1B受体两种亚型 [2]。AT1受体尤其AT1A在肾脏表达最丰富,ANGII的主要功能,如调节血压、肾血流量、肾小球滤过率、水和电解质代谢以及剌激组织细胞增生等主要是由AT1受体介导。有研究表明肾脏局部的RAS活化可引起肾小球硬化及系膜细胞肥大,而此作用亦是通过AT1活化实现的。AT1受体还与肾小管间质病变有关,肾间质成纤维细胞上也有丰富的AT1受体的表达,与AngII结合后,可刺激肾间质细胞产生胶原Ⅲ。
AT2受体:AT2受体介导的生理、病理功能尚未完全清楚。AT2受体在胚胎的各组织中均有广泛和丰富的表达,出生后大多数组织器官的AT2受体迅速减少或消失。最近的一些研究发现,在肾包膜、肾血管、球旁器以及肾小球系膜细胞也有少量AT2受体存在,AT2受体在成年人体肾组织中的含量,占肾组织中AngII受体总量的5%[4]。Muller等[5]的研究则发现,AT2受体也可以介导AngII引起的部分肾血管收缩作用,不过这种收缩作用较微弱,正常情况下被内源性一氧化氮(NO)的舒血管作用所抵消;只有用NO合成酶抑制剂(LNAME)抑制内源性NO的产生后,才可以显露出来。但也有不一致的结论,Arima[6]等在体外用微灌注法研究兔离体肾脏的入球小动脉时却发现,在用AT1受体拮抗剂(CV11974)拮抗AngII引起的血管收缩时,随灌注液AngII浓度的升高,入球小动脉反而扩张,这种扩张可被AT2受体拮抗剂抵消。提示,AT2受体介导舒血管作用。因此,大多数学者认为ATl受体和AT2受体对血管收缩和细胞生长的作用是相互拮抗的,但通常ATl受体起主导作用。AT2受体的研究,有助于进一步明确AngII的作用机制及其对肾脏的影响。
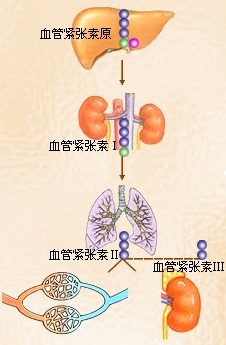
图1为肾素-血管紧张素系统
【血管紧张素2高的原因】
1.因失血引起循环血量减少或肾疾病导致肾血流量减少等,可促进肾小球旁器的球旁细胞分泌肾素,进入血液后,使血中由肝生成的血管紧张素原水解为血管紧张素ⅰ,它随血液流经肺循环时,受肺所含的转化酶作用,被水解为8肽的血管紧张素ⅱ,部分血管紧张素ⅱ受血浆和组织液中血管紧张素酶a的作用,被水解为7肽的血管紧张素ⅲ。
正常情况下,由于肾素分泌很少,血中血管紧张素也少,对血压调节不起明显作用。但当大失血时,由于动脉血压显著下降使肾血流量减少,血管紧张素生成增多,对防止血压过度下降而使血压回升却起重要作用。肾血管长期痉挛或狭窄的患者,因肾血流量减少,血管紧张素生成增多可导致肾性高血压。
2.血管紧张素主要是肾脏分泌的一种激素.它与肾上腺素有关.如果肾上腺素增多也会引起血管紧张素增高.
3.血溶量也有关.如果血溶量不足也会导致血管紧张素2升高;
4.某些肿瘤也可以分泌这种激素。
【AngII对肾脏的作用及其机制】
AngII是肾素-血管紧张素系统(RAS)中最主要的生物活性物质,在肾脏损害中作用重要。AngII与肾脏细胞膜上的血管紧张素Ⅰ型受体结合后,可通过血液动力学依赖和非血液动力学依赖两种途径造成肾脏损害,既可通过增加肾小球毛细血管内压这一间接途径引起肾脏的损害,也可通过刺激肾脏细胞分泌TGF-?等各种生长、细胞因子,直接造成肾脏损害。概括起来主要有以下几方面作用:
(1)缩血管作用:AngII是一种血管活性物质,它通过收缩大血管及肾小球小动脉及肾小球毛细血管网来调节肾小球内血液动力学的变化。它优先收缩肾小球出球小动脉的作用,使肾小球内压增高,引起肾小球损伤。
(2)收缩系膜细胞作用:AngII亦可收缩系膜细胞,影响肾小球内毛细血管滤过屏障,引起肾小球超滤系数下降。
(3)对肾小管的作用:AngII影响肾小管的Na+-H+,转换系统和Na+/HC03+的共同转运,从而影响近端肾小管的Na+、HCO3+的分泌。AngII不仅调控酸硷平衡以及细胞外液的重要转运系统,而且促进肾小管上皮的产氨基作用,使NH3较多地分泌到管腔。同时还对细胞增殖起重要作用。AngII促使该系统的转运,一方面调节了酸碱和体钠平衡,同时调节了该部分小管的肥大、增生。肾小管及其附近RAS过度活跃,有可能使小管细胞肥大加剧,此外AngII促进肾小管上皮的产氨基作用,能直接促进肾脏肥大,亦能通过激活补体引起C569增加,后者作为膜攻击复合物引起肾组织损伤。此外,AngII促进肾小管上皮细胞钠重吸收增加,增加了肾组织氧耗,引起肾组织氧相对不足,这些都是肾脏疾病慢性进展的重要机制。
(4)促肾生长因子作用:近年来研究发现AngII还是一种促肾生长因子,对细胞增殖起重要作用。它不仅可直接促进肾小球系膜细胞增生和肥大,还能刺激系膜细胞、上皮细胞、间质纤维母细胞分泌生长因子和细胞因子等,如TGF-?、PDGF、CTGF、IL-6、TNFa、 MCP-1、 PA1、金属蛋白酶等,导致肾小球系膜细胞增生和细胞外基质积聚,促进炎症细胞在肾小球和肾小管间质的浸润,促进肾小球和肾小管的纤维化。AngII能刺激一些细胞因子生成,尤其是TGF-1的作用引起了很多学者的兴趣,大多数动物模型中均证实AngII是诱发TGF-1表达的重要因素,给予ACEI或ATl受体拮抗剂可明显抑制TGF-1产生并延缓肾功能下降。ACE引起TGF-?1增多的机理为AngII与ATl受体结合后可激活蛋白激酶(PKC),PKC再通过一系列细胞内信号传导系统引起原癌基因如c-fos和c-Jun表达,二者形成AP-l样转录因子,己知TGF-?1基因启动子上存在AP-l样转录位点,因而可诱导TGF-?1表达。因而AngII亦可通过TGF-?1介导促进ECM积聚,另外,AngII还能抑制ECM降解,除了它能借助于TGF-1作用之外,AngII可直接上调PAI-ImRNA表达。有学者在肾小球硬化的动物模型实验中观察到,苯那普利通过阻断肾内RAS,在下调肾组织TGF-?1表达和细胞外基质积聚中起重要作用。
(5)AngII对巨噬细胞的作用:①巨噬细胞上有ArlgII的受体,AngII可以直接激活巨噬细胞;②AngII诱导单核/巨噬细胞释放趋化因子,灌注AngII可以引起间质性肾炎;③ 激肽系统可能参与这一过程,ACEI能增加缓激肽,刺激前列腺素或NO的产生,反过来降低单核/巨噬细胞的粘附和趋化。ACEI治疗后大鼠肾内激肽释放酶mRNA表达增加,可被特异性B2型缓激肽受体拮抗剂拮抗,用抑肽酶减少激肽降解可使尿蛋白排出减少,表明ACEI治疗后缓激肽水平增加与尿蛋白减少相关。故目前认为缓激肽在肾脏疾病的进展中也起着重要的作用。
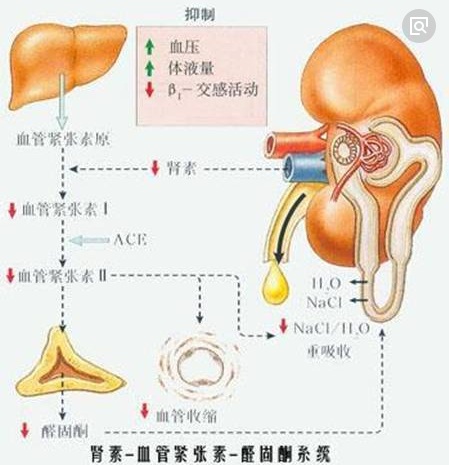
图2为肾素-血管紧张素-醛固酮系统
【主要参考资料】
[1]http://club.xywy.com/static/20160529/108925811.htm.
[2]http://wenda.so.com/q/1449101817721776?src=140.
[3]http://www.haodf.com/zhuanjiaguandian/zhws65415_90155.htm.