


Evolutionary convergence in the biosyntheses of the imidazole
Nov 5,2019
The imidazole group is an ubiquitous chemical motif present in several key types of biomolecules. It is a structural moiety of purines, and plays a central role in biological catalysis as part of the side-chain of histidine, the amino acid most frequently found in the catalytic site of enzymes. Histidine biosynthesis starts with both ATP and the pentose phosphoribosyl pyrophosphate (PRPP), which is also the precursor for the de novo synthesis of purines. These two anabolic pathways are also connected by the imidazole intermediate 5-aminoimidazole-4-carboxamide ribotide (AICAR), which is synthesized in both routes but used only in purine biosynthesis. Rather surprisingly, the imidazole moieties of histidine and purines are synthesized by different, non-homologous enzymes. As discussed here, this phenomenon can be understood as a case of functional molecular convergence.

Introduction
The imidazole group is a five-membered heterocyclic chemical compound containing a tertiary nitrogen and an imino group. It is present in the structure of several key molecules of major biological significance, most notably purines and histidine. The structure of the unsubstituted imidazole ring has two nitrogen atoms in positions 1 and 3 (also named pyrrole- and pyridine-nitrogen atoms, respectively) (Fig 1). The pyrrole nitrogen is a very weak acid with a pKa = 14.4. In contrast, the pKa of the pyridine nitrogen is 6.9, which allows it to accept a proton under neutral pH conditions [1]. This phenomenon does not occur in the imidazole moiety of purines, but it takes place in the imidazole side-chain of histidine, whose pKa value is in the range of 6 to 7 as a result of the electron-withdrawing inductive effect of the protonated amino group [2,3]. Bioinformatic analyses have shown that histidine is the most frequently found catalytic residue in the active sites of enzymes [4]. This phenomenon may be explained by the level of ionization of the imidazole moiety of histidine, which is estimated to be between 9–50%, resulting in its well-known ability to act as a general acid or base to donate or accept protons during a chemical reaction. This property, together with the stable complexes that it can form with different metallic cations such as Cu2+, Co2+, Zn2+, and Mn2+ [1,5,6], plays a key role in our understanding of the catalytic propensity of histidine.
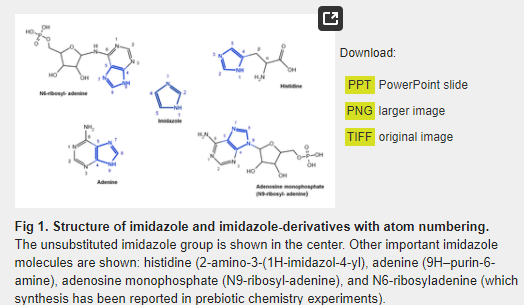
Abiotic synthesis of adenine and histidine
Adenine was the first imidazole-bearing molecule synthetized under abiotic conditions from an aqueous solution of ammonium cyanide (NH4CN) [7]. Since then, many other purines have also been synthesized in prebiotic simulations, including guanine and xanthine [8], hypoxanthine [9], 8-hydroxymethyladenine [10], and 2,6-diaminopurine [11]. Outstandingly, the imidazole group, as well as its 2-methyl derivative were obtained by Oró et al. [12] using glyoxal, aldehydes and ammonia as precursors.
The first attempt to synthesize histidine under possible prebiotic conditions was reported by Shen et al. [13,14]. In the proposed reaction, D-erythrose and formamidine are condensed, leading to imidazole-4-acetaldehyde, which then forms histidine via a Strecker synthesis with a relatively high yield of 3.5% based on the ratio of His/erythrose (Fig 2). As argued below, a detailed examination of this reaction suggests that the conditions of synthesis are prebiotically unrealistic, and that it is unlikely it took place in the primitive Earth.
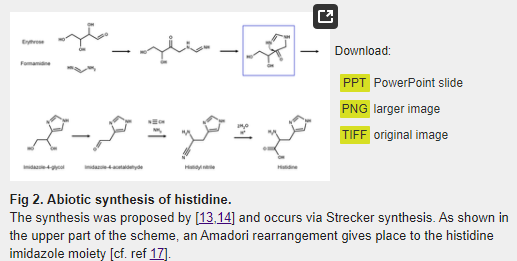
Even though the a priori selection of the D-enantiomer of erythrose is irrelevant since the stereocenters do not partake in the synthesis, and the reaction would work if other 4-carbon aldoses were used as precursors, D-erythrose and D-threose are minor, unstable products of the formose reaction [15], and therefore could not have been available in the concentration required for the reaction reported by Shen et al. [13,14] to take place. In addition, formamidine is a labile compound that is rapidly hydrolyzed into formic acid and ammonia [16], which also makes it very difficult to achieve the concentration required for the proposed reaction (0.3 M). As concluded elsewhere, all this data strongly hinders the prebiotic significance of this synthesis [17].
Nevertheless, it is quite interesting that in the reaction mechanism proposed by Shen et al. [13,14] the formation of the imidazole group is the product of an Amadori rearrangement (Fig 2), which is also the mechanism involved in the synthesis of the imidazole moiety of imidazole glycerol phosphate (IGP) by the IGP synthase (HisHF) enzyme during histidine biosynthesis. However, as argued elsewhere [17], all the evidence suggests that there is no direct evolutionary connection between the abiotic synthesis reported by Shen et al. [13,14] and the extant histidine anabolism.
As of today, no imidazole-bearing amino acid has been produced in Miller-Urey-type experiments or in other type of laboratory simulations, and the search of histidine and its degradation products in carbonaceous chondrites has also yielded negative results. These results suggest that histidine as such may have not been present in the prebiotic environment [17–19] but, as argued here, may be in fact a product of very early biological evolution.
Role of imidazolides in the evolution of catalysis
Although it has been shown that clays, metallic cations, and relatively simple compounds like proline and other amino acids can affect the rates of chemical reactions under possible primordial conditions, the emergence of biological catalysis remains one of the key questions in modern biology.
The discovery of the catalytic properties of RNA [20,21] provided support for the RNA world proposal, where it is assumed that RNA molecules played a key role in both heredity and catalysis during the very early stages of biological evolution.
Although how the RNA world evolved from the prebiotic environment remains an open issue, there have been advances in our understanding of the role that imidazole and imidazole-derivatives could have played during this evolutionary stage. For instance, it has been demonstrated that 2-aminoimidazole can act as an activating group for non-enzymatic RNA copying by a mechanism involving an imidazolium-bridged dinucleotide intermediate at pH = 7 [22]. Unsubstituted imidazole may have also played a role during the RNA world, as suggested by the demonstration that RNA cleavage can be catalyzed by imidazole in buffer [23]. The unsubstituted imidazole group can also act as a cofactor for the hepatitis delta virus (HDV) self-cleaving ribozyme [24]. The reaction mechanism of this ribozyme involves a nucleophilic attack of the 2′-OH on the phosphorus in the phosphodiester backbone, where the catalytic cytosine accepts the proton from the attacking 2′-hydroxyl group. As shown by Perrotta et al. [24], the incubation with unsubstituted imidazole buffer (200 mM, pH 7.4) restores the catalytic activity of a mutated HDV ribozyme. This shows that imidazole by itself can act as a co-catalyst in reactions involving RNA molecules, which could be of key significance in an RNA world scenario, and of considerable significance in our understanding of the early evolution of biological catalysis.
The catalytic activity of His-containing di- and tri-peptides has also been investigated. Histidyl-histidine (His-His) catalyzes the dephosphorylation of deoxyribonucleoside monophosphate, the hydrolysis of oligo (A)12, and the oligomerization of 2',3'-cAMP under cyclic wet-dry reaction conditions [25]. More recent studies have shown that seryl-histidine (Ser-His) catalyzes the oligomerization of trimers of imidazole-activated nucleotides [26,27], and catalyze peptide bond formation between two amino acids, an activity also found in seryl-histidyl-glycine (Ser-His-Gly) [28,29].
Evolution of enzyme catalytic activity from an RNA world
Although there have been alternative proposals to explain the origin of ribosome-mediated protein synthesis based on the simultaneous appearance of both RNA and proteins [30,31], it can also be argued that the ribosome first appeared as an evolutionary outcome of the interaction of prebiotically synthesized amino acids with catalytic RNA molecules. How enzymes first evolved remains an open question, but it is reasonable to assume that their active sites must be one of their most conserved portions. However, if histidine was absent in the primitive Earth, how can its ample distribution in the active sites of enzymes be explained?
In what may be the first attempt to explain the origin of histidine biosynthesis, over 40 years ago Harold White III correlated the start of the histidine biosynthetic pathway, which involves a condensation reaction between ATP and the sugar phosphoribosyl pyrophosphate (PRPP), with the possibility of an ancient metabolism mediated by nucleic acid enzymes [32]. In his evolutionary biochemistry approach, White proposed that histidine was, in fact, the molecular vestige of an ancient catalytic nucleotide that was part of the RNA world [32,33]. According to White, this possibility is reinforced by a) the well-known fact that the histidine anabolic pathway is connected with the de novo synthesis of purines by the usage of PRPP and the 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR) intermediate; b) His is the only known amino acid with a ribonucleotide-starting biosynthesis; and c) histidine is the only imidazole-bearing amino acid.
While it is true that histidine is the only biological amino acid with an imidazole side chain, the other two issues merit a reexamination. As underlined by White [32], during the first step of the histidine biosynthesis, PRPP, whose synthesis in prebiotic conditions has been recently reported by Akouche et al. [34], is condensed with a molecule of ATP to form N’5- phosphoribosyl -ATP (PRATP) (Fig 3). Quite surprisingly, however, the imidazole component of the ATP does not partake in the biosynthesis of the imidazole group of histidine. Instead, the histidine’s imidazole moiety is biosynthesized de novo by a mechanism completely different from that of purines.
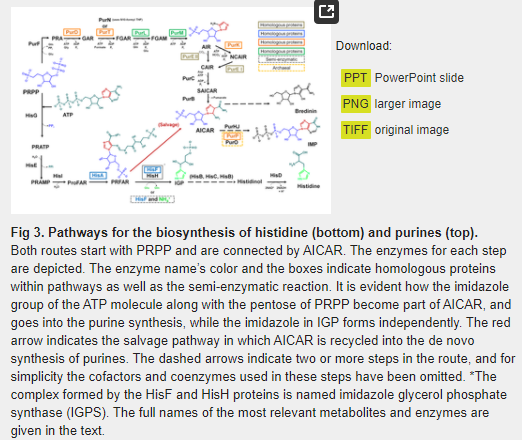
As shown on Fig 3, histidine- and purine biosyntheses are indeed connected by the imidazole intermediate AICAR, which is formed independently in the two pathways. In the purine route it is formed from succinyl-AICAR (SAICAR), and from N'-((5'-phosphoribulosyl) formimino)-5-aminoimidazole-4-carboxamide-ribonucleotide (PRFAR) in the histidine pathway, but it does not play any further role in the subsequent steps of the histidine anabolism. Instead, it is recycled directly into the purine pathway, where it is used as a substrate by the bifunctional enzyme AICAR transfromylase/inosine monophosphate (IMP) cyclo-hydrolase (PurHJ) [EC: 2.1.2.3/EC: 3.5.4.10], that catalyzes the final two steps in the route and synthesizes IMP (Fig 3). We suggest that the incorporation of AICAR from the histidine biosynthetic route into the de novo synthesis of purines can be best understood as an intracellular salvage pathway (Fig 3), in which the imidazole compound is used to supply the requirements of purine synthesis.
Results
Imidazole-biosynthetic enzymes HisF and PurM: A case of evolutionary convergence
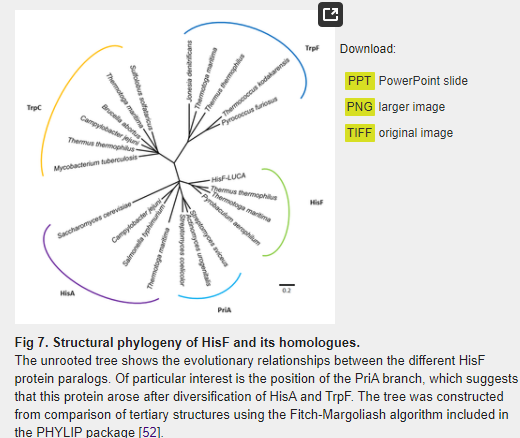
The E. coli PurM (354 aa) and HisF (258 aa) proteins share only 12.2% of overall sequence identity (21.4% similarity) (S1 Fig). When the crystallographic structures of the two proteins are superimposed, the root-mean-square deviation (RMSD) of the α-carbons is 8.27 Å (Fig 6A). This very high RMSD value reflects of the very different domain architectures of the two enzymes. As noted above, HisF has the (β/α)8-barrel fold and belongs to the ribulose-phoshate binding barrel superfamily, while PurM adopts a completely different fold and is part of the PurM superfamily. Considering these structural differences, it can be readily concluded that these are different enzymes with different evolutionary histories that share no common ancestor. The evolutionary relationship between HisF and its structural homologues is depicted in the structural phylogeny shown in Fig 7. The branches more closely related to HisF are those corresponding to the enzyme N’-[(5′-phosphoribosyl) formimino]-5-aminoimidazole-4-carboxamide ribonucleotide (ProFAR) isomerase [EC:5.3.1.16] (HisA). It has been shown that HisF and HisA share a similar internal organization that can be explained as two paralogous modules half the size of the extant sequences [54]. An ancestral module duplicated and gave rise to the ancestral hisA gene which, in turn, underwent another duplication and fused, giving rise to the hisF gene [35,54,55]. This shared evolutionary history explains the closeness between the branches.
- Related articles
- Related Qustion
- Imidazole: A Versatile Molecule in Chemistry Nov 5, 2024
Imidazole is a significant compound in the field of chemistry, known for its wide range of applications and unique properties.
- General Description of Imidazole Feb 11, 2022
Imidazole is a planar, five-membered, unsaturated, 6π electron ring system, comprised of three carbon atoms and two nitrogen atoms at 1,3-positions with two olefinic bonds.
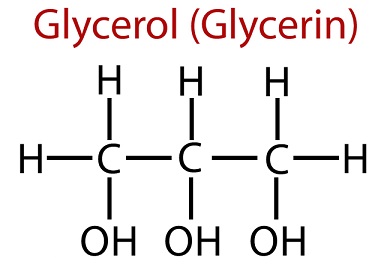
Glycerol is most commonly used for constipation, improving hydration and performance in athletes, and for certain skin conditions. It is also used for meningitis, stroke, obesity, ear infections, and other conditions, but there is no good s....
Nov 5,2019API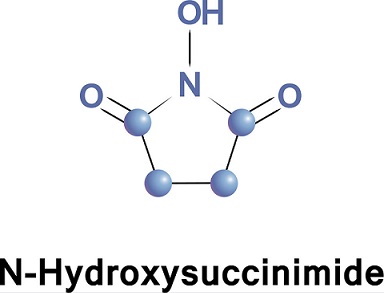
In contrast to N-hydroxyphthalimide, N-hydroxysuccinimide5 is water soluble, and we have found that acylamino acid esters of this compound meet the important requirements of crystallinity and high reactivity. Consequently we have undertake....
Nov 5,2019Biochemical Engineering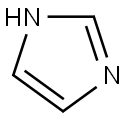
Imidazole
288-32-4You may like
- Imidazole
-

- $0.00 / 25KG
- 2025-12-15
- CAS:288-32-4
- Min. Order: 1KG
- Purity: 99%
- Supply Ability: 1000mt/year
- Imidazole
-

- $6000.00 / 1000kg
- 2025-12-15
- CAS:288-32-4
- Min. Order: 1000kg
- Purity: 99%
- Supply Ability: 500 tons
- Imidazole
-

- $6000.00 / 1000kg
- 2025-12-15
- CAS:288-32-4
- Min. Order: 1000kg
- Purity: 99%
- Supply Ability: 500 tons