



Ibrutinib
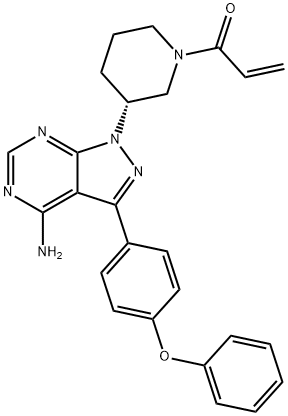
- CAS No.
- 936563-96-1
- Chemical Name:
- Ibrutinib
- Synonyms
- Lbrutinib;1-[(3R)-3-[4-Amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl]-1-piperidinyl]-2-propen-1-one;PCI-32765;Ibrutinib (PCI-32765);IBRUTINIB (PCI-32765);PCI32765;1-{3-[4-AMino-3-(4-phenoxy-phenyl)-pyrazolo[3,4-d]pyriMidin-1-yl]-piperidin-1-yl}-propenone;(R)-1-(3-(4-Amino-3-(4-phenoxyphenyl)-1H-pyrazolo-[3,4-d]pyrimidin-1-yl)piperidin-1-yl)prop-2-;(R)-1-(3-(4-aMino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyriMidin-1-yl)piperidin-1-yl)prop-2-en-1-one;CS-188;Ibutinib
- CBNumber:
- CB12515873
- Molecular Formula:
- C25H24N6O2
- Molecular Weight:
- 440.5
- MDL Number:
- MFCD20261150
- MOL File:
- 936563-96-1.mol
- MSDS File:
- SDS
| Melting point | 153-158°C |
|---|---|
| Boiling point | 715.0±60.0 °C(Predicted) |
| Density | 1.34 |
| storage temp. | -20°C |
| solubility | Soluble in DMSO ( up to at least 25 mg/ml) |
| form | solid |
| pka | 4.09±0.30(Predicted) |
| color | White or off-white |
| Stability | Stable for 1 year from date of purchase as supplied. Solutions in DMSO may be stored at -20°C for up to 3 months. |
| InChIKey | XYFPWWZEPKGCCK-GOSISDBHSA-N |
| SMILES | C(N1CCC[C@@H](N2C3C(C(C4=CC=C(OC5=CC=CC=C5)C=C4)=N2)=C(N)N=CN=3)C1)(=O)C=C |
| CAS DataBase Reference | 936563-96-1 |
| FDA UNII | 1X70OSD4VX |
| NCI Drug Dictionary | ibrutinib |
| ATC code | L01EL01 |
SAFETY
Risk and Safety Statements
| Symbol(GHS) |   GHS07,GHS06 |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Signal word | Danger | |||||||||
| Hazard statements | H360-H319-H315-H335-H302 | |||||||||
| Precautionary statements | P264-P280-P302+P352-P321-P332+P313-P362-P264-P270-P301+P312-P330-P501-P264-P280-P305+P351+P338-P337+P313P | |||||||||
| Hazardous Substances Data | 936563-96-1(Hazardous Substances Data) | |||||||||
| NFPA 704 |
|
Ibrutinib price More Price(42)
| Manufacturer | Product number | Product description | CAS number | Packaging | Price | Updated | Buy |
|---|---|---|---|---|---|---|---|
| Cayman Chemical | 16274 | Ibrutinib ≥98% | 936563-96-1 | 1mg | $37 | 2024-03-01 | Buy |
| Cayman Chemical | 16274 | Ibrutinib ≥98% | 936563-96-1 | 5mg | $108 | 2024-03-01 | Buy |
| Cayman Chemical | 16274 | Ibrutinib ≥98% | 936563-96-1 | 10mg | $178 | 2024-03-01 | Buy |
| Cayman Chemical | 16274 | Ibrutinib ≥98% | 936563-96-1 | 50mg | $793 | 2024-03-01 | Buy |
| Tocris | 6813 | Ibrutinib ≥98%(HPLC) | 936563-96-1 | 10 | $150 | 2021-12-16 | Buy |
Ibrutinib Chemical Properties,Uses,Production
Bruton tyrosine kinase (BTK) inhibitor
Ibrutinib[936563-96-1] is a Bruton's tyrosine kinase (BTK) inhibitor used for the treatment of chronic lymphocytic leukemia (CLL) and mantle cell lymphoma (MCL). MCL and CLL are both B-cell non-Hodgkin lymphomas, characterized by treatment resistance and high rate of relapse. Conventional chemotherapy and immunotherapy lack specificity and often result in grade 3 or 4 adverse reactions. Ibrutinib selectively binds to BTK, an essential component for B lymphocyte formation, differentiation, signaling, and survival, irreversibly inhibiting its activity. It effectively inhibits tumor cell proliferation and survival. Additionally, it is rapidly absorbed after oral administration, reaching peak blood concentration in 1-2 hours, and adverse reactions are usually grade 1 or 2. Ibrutinib will become a new treatment option for CLL and MCL.
On November 13, 2013, the US Food and Drug Administration (FDA) accelerated the approval of Imbruvica (generic name: Ibrutinib) manufactured by Pharmacyclics and Johnson & Johnson for the treatment of a rare aggressive blood cancer called mantle cell lymphoma (MCL). Ibrutinib is an orally available Bruton's tyrosine kinase (BTK) inhibitor, which is a first-in-class innovative drug. It was granted breakthrough therapy designation by the FDA in February 2013 and approved for the treatment of MCL and chronic lymphocytic leukemia (CLL) on November 13, 2013, and February 12, 2014, respectively. This drug selectively and irreversibly inhibits BTK by covalently binding to the active site cysteine residue (Cys-481) of the target protein Btk, effectively preventing the tumor from migrating from B cells to lymphoid tissues that are conducive to tumor growth.
Information was collated by Xiaonan editor of Chemicalbook.
Research and development process
Ibrutinib(936563-96-1) was developed by American Celera Genentech (Celera Genomics) that was well known because it was the first one drawn "human genome". Pan Zhengying (now Beijing University Shenzhen Graduate School Distinguished Fellow) as the first author of this research and published the process of Ibrutinib in 2007 (ChemMedChem 2 (1): 58-61). But Celera had transferred the development rights to Pharmacyclics Company of California due to funding and resources. Although Pharmacyclics Company was in a very difficult stage, only $ 0.64 for the stock, facing delisting and bankruptcy in that time, company's management has managed to raise funds, spent only $ 2 million to down payment, 100 million shares and future sales royalty and milestone payment of royalties more to get Ibrutinib. In 2011, Johnson & Johnson subsidiary Janssen pay $ 150 million upfront payment in order to get the right to cooperate with Pharmacyclics. Once the new drugs in clinical trials and approved to listing, Pharmacyclics company will get $ 975 million in total revenue, and they will share the sales.When the deal was announced, Pharmacyclics stock is only $ 12. Market value was only several hundred million dollars. However, the stock is already $ 123, $ 9.05 billion value now.
Mechanism
Signaling pathway of B cell antigen receptor (BCR) is a key driver of tumor growth and spread. BTK as an indispensable participant for BCR signal peptide, it is very important for formation, differentiation, messaging and survival of B lymphocyte. BTK is an identifiable signal peptide molecules of BCR channel. When the signal peptide molecules across the B lymphocyte surface receptors, required channel is activated for transportation, chemotaxis and adhesion, which provide a convenience to be B-cell malignancies.
Ibrutinib is a small molecule can selectively and covalently bound to a cysteine residue (Cys-481) BTK active site, and irreversibly inhibit BTK activity, thereby inhibiting BCR activated signaling pathway, which could effectively prevent the tumor from B cells to lymphoid tissues where suitable for tumor growth, to reduce B cell proliferation and induce apoptosis of malignant cells, which play a role in the treatment of CLL and MCL. Non-clinical studies have shown that Ibrutinib can inhibit malignant B lymphocyte proliferation and survival.
Metabolism and Elimination
Ibrutinib(936563-96-1) primarily metabolized by the cytochrome P450 (CYP3A and a small part of the CYP2D6) and produce a variety of metabolites after metabolism. The metabolite (PCI-45227) is a kind of dihydrodiol substance which is an activity of inhibiting BTK.Compared with Ibrutinib, PCI-45227 has much stronger inhibition on BTK, which is about 15 times stronger. At steady state, the average rate of metabolism of PCI-45227 is around 1~2.8.
Apparent clearance (CL/F) of Ibrutinibis is approximately 1 000 L/h, the half-life (t1/2) is 4~6 h. Ibrutinib mainly be metabolites in the body, excrete with the feces. To healthy subjects by oral radioactive 14C-labeled Ibrutinib, found that nearly 90% of the radiation were eliminated in 168 h, most (about 80%) of them were excreted with the feces, and nearly 10% were excreted in the urine, about 1 % be prototypes were excreted with the feces.
Elimination of Ibrutinib will not make a difference in age (37 to 84 years) and gender, but systemic exposure of patients with moderate hepatic injury will be 6 times higher than in healthy subjects.
Patents
Foreign patents: WO 2008039218 (compound); WO 2013003629 (purposes)
US Patent Number: 7,514,444, 7,718,662, patents is valid: December 2026
Domestic patent: CN101610676A, CN101610676B, CN101805341A, CN101805341B, CN102746305A, CN102887900A
Originator
Celera/Pharmacyclics (United States)
Uses
Ibrutinib is a highly selective Bruton’s tyrosine kinase (Btk) irreversible inhibitor.
Definition
ChEBI: Ibrutinib is a member of the class of acrylamides that is (3R)-3-[4-amino-3-(4-phenoxyphenyl)pyrazolo[3,4-d]pyrimidin-1-yl]piperidine in which the piperidine nitrogen is replaced by an acryloyl group. A selective and covalent inhibitor of the enzyme Bruton's tyrosine kinase, it is used for treatment of B-cell malignancies. It has a role as an EC 2.7.10.2 (non-specific protein-tyrosine kinase) inhibitor and an antineoplastic agent. It is a pyrazolopyrimidine, an aromatic amine, an aromatic ether, a member of acrylamides, a N-acylpiperidine and a tertiary carboxamide.
Indications
Ibrutinib is a non-receptor Bruton's tyrosine kinase inhibitor approved for the treatment of relapsed chronic lymphocytic leukemia(CLL), mantle cell lymphoma (MCL) and WaldenstrÖm's Macroglobulinemia (WM).
brand name
Imbruvica
Side effects
The more common side effects of Ibrutinib are diarrhoea and bleeding. Therefore, you should drink plenty of water during treatment, in order to reduce the risk of dehydration using diarrhoea. Bleeding problems can be serious and may even lead to death. This may be manifested by blood or black stools (that look like tar); pink or brown urine; accidental bleeding, or severe or uncontrollable bleeding; spitting up blood or vomit that looks like coffee grounds; coughing up blood; bruising or small red or purple spots on the skin; dizziness; confusing changes in speech; or a headache that lasts for a long time or is severe. Prompt medical attention is needed if a serious condition develops.
Other possible side effects are: increased risk of infection, fever, chills, weakness, confusion, or other signs or symptoms of infection; increased risk of heart disease, rapid and irregular heartbeat; dizziness; lightheadedness; shortness of breath; swelling of the feet, ankles, or legs; discomfort in the chest; or feeling lightheaded. High blood pressure, reduced blood counts, second primary cancer and tumour lysis syndrome (TLS). Different disease treatments exhibit different adverse effects, so the above side effects may not occur in full.
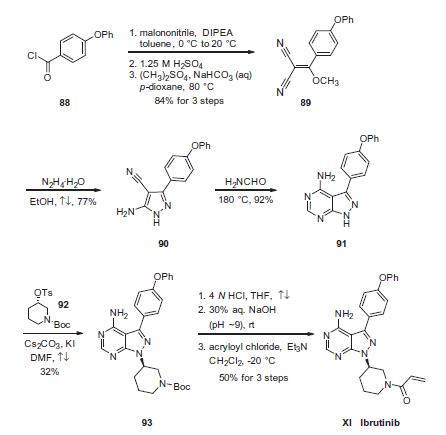
Synthesis
Condensation of commercially available 4-phenoxybenzoyl chloride (88) with malononitrile followed by acidic quench and O-methylation with dimethyl sulfate furnished vinyl dinitrile 89 in 84% yield over the three-step sequence. Next, treatment with hydrazine hydrate in refluxing ethanol secured aminopyrazole 90 and this was followed by treatment with neat formamide at elevated temperature to furnish pyrimidopyrazole 91 in excellent conversion. Selective alkylation of the pyrazole nitrogen with commercially- available (S)-piperidinyl tosylate (92) proceeded in 32% yield. Finally, liberation of the amide followed by pH adjustment and amide bond formation with acrolyl chloride furnished ibrutinib (XI) in 50% over the three-step sequence.
Metabolism
Ibrutinib is metabolised primarily by CYP3A4 to
produce a dihydrodiol metabolite with an inhibitory
activity towards BTK approximately 15 times lower
than that of ibrutinib. Involvement of CYP2D6 in the
metabolism of ibrutinib appears to be minimal.
After a single oral administration of radiolabeled
[14C]-ibrutinib in healthy subjects, approximately 90%
of radioactivity was excreted within 168 hours, with the
majority (80%) excreted in the faeces and <10% in the
urine. Unchanged ibrutinib accounted for approximately
1% of the radiolabeled excretion product in faeces and
none in urine.
storage
+4°C
Mode of action
Ibrutinib is the second oral agent approved for the treatment of MCL. It works by irreversibly inhibiting Bruton’s tyrosine kinase (Btk) leading to the inhibition of B-cell receptor signaling and resulting in the reduction of malignant B-cell proliferation and induction of cell death. Btk plays an important role in the differentiation, development, proliferation, and survival of B cells via activation of cell-cycle regulators and regulating the expression of pro- and antiapoptotic proteins. Aberrant Btk activity results in a variety of B-cell malignancies including MCL. Ibrutinib inhibits Btk by irreversibly binding to cysteine-481 in the active site thereby inhibiting phosphorylation of tyrosine-223 and affecting downstream B-cell signaling pathways.
References
1) Honigberg?et al.?(2010),?The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy;?Proc. Natl. Acad. Sci. USA?107?13075 DOI:10.1073/pnas.1004594107
2) Ponader?et al.?(2012),?The Bruton tyrosine kinase Inhibitor PCI-32765 thwarts chronic lymphocytic leukemia cell survival and tissue homing in vitro and in vivo;?Blood?119?1182
DOI:10.1182/blood-2011-10-386417
3) De Rooij?et al.?(2012),?The clinically active BTK inhibitor PCI-32765 targets B-cell receptor- and chemokine-controlled adhesion and migration in chronic lymphocytic leukemia;?Blood?119?2590
DOI:10.1182/blood-2011-11-390989
4) Pavlasoca?et al.?(2016),?Ibrutinib inhibits CD20 upregulation on CLL B cells mediated by the CXCR4/SDF-1 axis;?Blood?128?1609 DOI:10.1182/blood-2016-04-709519
5) Sagiv-Barfi?et al.?(2015),?Therapeutic antitumor immunity by checkpoint blockade is enhanced by ibrutinib, an inhibitor of both BTK and ITK; Proc. Natl. Acad. Sci. USA?112?E966 DOI:10.1073/pnas.1500712112
6) Weber?et al.?(2017),?Bruton's Tyrosine Kinase: An Emerging Key Player in Innate Immunity,?Front. Immunol.?8?1454 DOI:10.3389/fimmu.2017.01454
Ibrutinib Preparation Products And Raw materials
| Supplier | Tel | Country | ProdList | Advantage | |
|---|---|---|---|---|---|
| AFINE CHEMICALS LIMITED | +86-0571-85134551 | sales@afinechem.com | China | 15222 | 58 |
| Handan Tongyi New Material Technology Co., Ltd | +8617330042575 | ty003@handantongyi.com | China | 351 | 58 |
| SyncoZymes (Shanghai) Co., Ltd., | +8613681683526 | lchen@syncozymes.com | China | 188 | 58 |
| Qiuxian Baitai New Material Co., LTD | +8618330912755 | sale2@hbyalin.com | China | 1658 | 58 |
| Shanghai Likang New Materials Co., Limited | +86-16631818819 +86-17736933208 | 3684455296@qq.com | China | 9300 | 58 |
| BEIJING SJAR TECHNOLOGY DEVELOPMENT CO., LTD. | +86-18600796368 | sales@sjar-tech.com | China | 485 | 58 |
| Hebei Chuanghai Biotechnology Co., Ltd | +8617732866630 | abby@chuanghaibio.com | China | 8773 | 58 |
| RongNa Biotechnology Co.,Ltd | +86-86-13583358881 +8618560316533 | Brad@rongnabiotech.com | China | 3060 | 58 |
| Jiangsu Magic Biotechnology Co., Ltd | +undefined13921770081 | SVP01@magicbiotech.cn | China | 92 | 58 |
| Henan Bao Enluo International TradeCo.,LTD | +86-17331933971 | deasea125996@gmail.com | China | 2472 | 58 |
Related articles
- BTK inhibitor Ibrutinib:Brand names,Uses,Dietary instructions and Toxicity
- Ibrutinib is a small molecule with the ability to form a covalent bond with Cys-481 in the ATP binding domain of BTK. The IC50....
- Jul 30,2025
- Immunosuppressant Ibrutinib:Indications, Class and Drug interactions
- Ibrutinib is a first-in-class, potent, orally administered covalently-binding inhibitor of BTK, it may stop the growth of canc....
- Mar 25,2025
- Ibrutinib: Uses, Mechanism of action and Side effects
- Ibrutinib is an oral, potent, selective and irreversible small molecule inhibitor of Btk. It is a prescription drug with the t....
- Apr 2,2024
View Lastest Price from Ibrutinib manufacturers
| Image | Update time | Product | Price | Min. Order | Purity | Supply Ability | Manufacturer | |
|---|---|---|---|---|---|---|---|---|
 |
2025-08-01 | Ibrutinib
936563-96-1
|
US $1.00 / g | 1g | 99% | 100kg | RongNa Biotechnology Co.,Ltd | |
 |
2025-08-01 | Ibrutinib
936563-96-1
|
US $0.00-0.00 / g | 1g | 99% HPLC | 10000 | HangZhou RunYan Pharma Technology Co.,LTD. | |
 |
2025-08-01 | Ibrutinib
936563-96-1
|
US $0.00 / g/Bag | 25g | 97%-103% | 10KGS | WUHAN FORTUNA CHEMICAL CO., LTD |
936563-96-1(Ibrutinib)Related Search:
1of4