阿哌沙班是直接因子Xa(factor Xa, FXa)抑制剂,作为抗凝剂,用于降低非瓣膜性房颤患者中风和全身性栓塞的风险,也可用于预防深静脉血栓(deep venous thrombosis, DVT)的形成。阿哌沙班由辉瑞(Pfizer)与百时美施贵宝(Bristol-Myers Squibb,BMS)联合研发,于2011年5月18日获欧洲药物管理局(EMA)批准上市,之后于2012年12月28日获美国食品药品管理局(FDA)批准上市,于2012年12月25日获得日本医药品医疗器械综合机构(PMDA)批准上市,于2013年1月22日获得中国食品药品监督管理局(CFDA)批准上市。
阿哌沙班的销量在2018年超越了同靶点首个药物利伐沙班,更是一举获得2018全球小分子药物销售NO.1,如此成绩让业界惊叹!而在其背后,这个分子是如何得到的,其过程更为精彩!
1.药物靶点FXa
Xa因子是凝血级联反应中的一个蛋白水解酶,功能是将凝血酶原转变为凝血酶,它的重要性在于通过内在机制或外来刺激引发凝血过程的最后环节。FXa作为酶分子具有放大效应,一个FXa分子可以将上万个凝血酶原经水解而激活成凝血酶,因而, FXa抑制剂也被认为是更有效率的抗血栓药。FXa是胰蛋白酶样的蛋白酶,直接抑制FXa的活性,从而抑制凝血酶的生成,阻断血栓形成和凝血。FXa不影响血小板功能,可降低出血的风险,在作用机制上优于凝血酶抑制剂和抗血小板聚集药物。
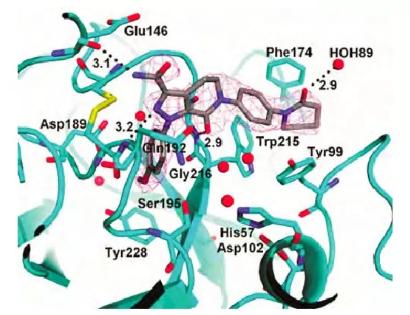
图1. FXa与阿哌沙班相互作用的X-ray
此外,我们来看看同靶点首个上市药物-利伐沙班。
利伐沙班,拜耳公司研发的一种口服药物,2008年在加拿大和欧盟率先上市,2009 年在我国上市,商品名为拜瑞妥®,用于选择性髋部或膝全替换手术病人VTE的预防等。
结构上,1998年拜耳公司开始组建团队开发小分子FXa抑制剂(FXa抑制剂最初源于水蛭),通过高通量筛选(数十万级别),得到苗头化合物结构,再通过一系列的优化,最终得到利伐沙班。
而进一步对比看伐沙班和阿哌沙班的结构,会发现阿哌沙班在结构上绝对不是很多传统同类靶点药物的me-too、me-better,而是经历了多种结构改造、骨架跃迁等操作,最终得到了新型FXa抑制剂。

图2. 利伐沙班&阿哌沙班的结构差异
2.从苗头化合物到候选化合物
从苗头到候选再到阿哌沙班,这个过程涉及多种原理和方法,如骨架跃迁、电子等排、分子拼合对接等,操作难度大、工作庞杂!前期工作与大部分药物发现的目的相同,即提高活性!
普筛→芳脒化合物
起初,多家公司报道了针对FXa的双芳香脒类抗血栓化合物,杜邦公司通过筛选IIb/IIIa受体拮抗剂(该公司曾深入研究)对FXa的活性,发现以下化合物对FXa的Ki值为38.5 μmol·L−1,但结构为单芳脒化合物。

图3. 普筛得到的单芳脒结构
单芳脒结构→双芳脒结构
在当时,双芳脒结构为主流结构,通过将右侧四氢异喹啉丁酮酸片段换成苯脒,同时进一步考察化合物对凝血酶(越强越好)和胰蛋白酶(越弱越好)的作用,探究选择性活性。
图4. 单芳脒到双芳脒结构的演变
双芳脒结构+异噁唑环取代
为降低分子柔性,将异噁唑环与酰胺之间的亚甲基除去,同时在异噁唑环上引入含羰基的侧链,但没有突破性进展。

图5. 引入含羰基侧链
双芳脒结构→联苯结构
逆“大流”,将双芳脒结构改为联苯,后发现,联苯基与FXa的结合腔S4是由Trp215、Tyr99和Phe174等氨基酸残基构成的疏水腔。分子模拟与FXa的结合特征表明,磺酰基的氧原子与Tyr99发生较强的氢键结合,且选择性很高。

图6. 引入联苯结构
联苯片段+近端苯环优化+侧链酯基变换
固定间位脒基、异噁唑环的乙酸甲酯基,考察中间的芳环对活性的影响;同时,由于结构中含有酯基,在体内会发生水解,在动物的血浆中有相当比例的游离酸存在,该游离酸对FXa的体内活性显著低于未水解的酯。

图7. 近端苯环优化+侧链酯基变换
异恶唑环→吡唑环
异噁唑啉不是芳香环,含有手性碳原子,同时证明5-位的碳链对活性影响不大,进而用吡唑替换异噁唑,活性未变,同时方便合成。后以吡唑结构为优化新起点(IC50已经达到0.13 nM)。

图8. 吡唑环的引入
吡唑-CH3 & -CF3化→活性&选择性
3-甲基取代的吡唑化合物,对FXa的抑制活性提高了1个数量级,3-三氟甲基取代的吡唑化合物,活性更强。多数化合物有较低的清除率分布容积和较长的半衰期,但口服生物利用度低于5%,未能达到口服给药的要求。分子结构中含有碱性较强的脒基(pKa为10.7),在体内分子带有正电荷,不易过膜吸收,因而口服吸收差。部分化合物PK/PD数据如下。

图9. 从活性扩展到更多药学性质
3.候选化合物的确定
DPC-423
苄胺代替苯脒结构,活性略弱于脒基化合物1-2个数量级,但因脒基具有pM水平的高活性,经得起活性的消减,以弥补过膜吸收性,优化药代+药效的总效果。
由于降低了分子的碱性,增加了过膜吸收性。在苄胺化合物中,吡唑环上有三氟甲基取代、联苯的近端苯环2位被氟取代、远端苯环4-甲磺酰基取代的化合物的口服生物利用度、半衰期长达7.5 h,用家兔动静脉分流血栓形成模型测定的ID50 = 1.1 μmol·kg−1·h−1,IC50 = 0.15 μmol·L−1。给予这些活性数据和药代动力学性质,杜邦公司确定了候选化合物,编号为DPC-423,进入开发。

图10. DPC-423化学结构
雷扎沙班
构效关系研究表明:DPC-423的甲磺酰基变换成氨磺酰基后,其活性未变,选择性提高了,但过膜性显著降低,系因增加了分子极性的缘故;远端苯环换成吡啶环,活性未变,过膜性仍较低;用咪唑、四氢吡咯或吗啉置换远端苯环,增加了助溶基团,提高了过膜性,但也因此与血浆蛋白的结合率>97%,也是活性降低的原因;为调整药代、维持活性强度和选择性,宜用1-(2-甲基咪唑)基取代;变换咪唑环上的取代基,羟甲基和氨甲基可能因极性强而降低活性;最终发现雷扎沙班(razaxaban),以盐酸盐形式进行临床研究,口服预防和治疗深度静脉栓塞疾病。

图11. 雷扎沙班化学结构
BMS740808
考虑到酰胺键可能在体内被水解成吡唑甲酸和联芳胺,后者具有潜在的致突变作用的风险,因而需要避免产生毒性的警戒结构,基于结构变换最小原则,将酰胺环合到吡唑环上,形成并合的杂环结构,以提高代谢稳定性。
将雷扎沙班的氨基苯并异噁唑与杂环并合吡唑连接,合成了一类新的化合物,构效关系总结如下:(1)变换远端苯环上的叔胺基,二甲胺甲基和3-R-羟基四氢吡咯活性最强,体内抗凝作用也最强。(2)在近端苯环的邻位引入氟原子,比相应的化合物活性弱。(3)3-羟基四氢吡咯R构型活性比其S构型强3倍,体内抗凝活性也强于S构型。(4)吡唑环上的三氟甲基用甲基代替,活性降低。比格犬的药代动力学研究表明,以下化合物具有的性质,即BMS740808。

图12. BMS740808化学结构
4.阿哌沙班的诞生
候选物为化合物DPC423、雷扎沙班、BMS740808,下一步是将这些片段作进一步的优化组合,从体外 FXa 活性、选择性、体内抗凝作用和药代性质等多维度进行优化,以确定更优结构。
将苯甲醚、吡唑并二氢吡啶酮、联苯叔胺等优势片段拼接起来,变换吡唑环上的取代基,合成系列化合物。其中化合物如甲磺酰基、氨甲酰基、氰基和四唑基等与三氟甲基化合物相比,都有较高的活性和选择性,体内抗凝作用显著强于BMS740808,提示这些基团可以移植在不同的母核上。相对比较,下图化合物具有良好的药代动力学性质、较低的清除率(CL = 0.32L·kg−1·h−1)、半衰期t1/2为5.6 h,口服生物利用度F=100%,因而将氨甲酰基固定,进一步优化。

图13. 氨甲酰结构的固定
在进行上述结构优化中,另一个同时研究的是探索远端苯环取代基的变换对活性影响,发现单苯环的对位N-甲基乙酰氨基取代是独特的,因为游离氨基或其他相关的基团活性都很差,提示,酰化的仲胺应是参与结合的重要基团。
氨甲酰基取代和候选物的确定纠正了三氟甲基过强的脂溶性,结合远端苯环对位N-甲基乙酰氨基取代,得到下述化合物,体外抑制FXa和体内抗凝血作用以及选择性都很强。对人体的蛋白酶和肝脏CYP的活性很弱,如远端苯环含内酰胺结构化合物与肝微粒体温孵的半衰期>100 min,Caco-2细胞的过膜性也非常高,血浆蛋白结合率为87%,家兔抗血栓的IC50=329 nmol·L−1,口服半衰期为5.8 h,口服生物利用度F=58%。终于,我们的小分子药王阿哌沙班,诞生了!

图14. 阿哌沙班终诞生
5.小结
阿哌沙班的分子结构设计过程,可谓精益求精!在早期获得多个活性非常非常好的化合物时,并未冒进,急于开发,而是继续对活性进行研究,以期为后续的成药性结构改造打下足够牺牲活性的“量”!
其结构改造的原理和方法看似广谱,实则表现出很赞的有机结合程度,尤其是跨越了利伐沙班的骨架结构以及早期苗头化合物的双脒结构,这种大胆放弃“经典结构”的举动对于国内药物分子的开发来说很具有指导意义。
参考资料:
1. Design and synthesis of isoxazolinederivatives as factor Xa inhibitors. J Med Chem.1999. DOI: 10.1021/jm980406a.
2. Discovery of 1-[3-(aminomethyl)phenyl]-N-[3-fluoro-2'-(methylsulfonyl)-[1,1'-biphenyl]-4-yl]-
3-(trifluoromethyl)-1H-pyrazole-5-carboxamide(DPC423), a highly potent, selective, and orally bioavailable inhibitor of blood coagulationfactor Xa. 2001. DOI: 10.1016/S0030-
4018(98)00117-5.
3. Structure-based design of novel guanidine/benzamidinemimics:potent and orally bioavailable factor Xa inhibitors as novel anticoagulants. 2003. DOI: 10.1021/jm020578e.
4. Discovery of 1-(4-methoxyphenyl)-7-oxo-6-(4-(2-oxopiperidin-1-yl)phenyl)-4,5,6,7-tetrahydro-1H-pyrazolo[3,4-c]pyridine-3-carboxamide(Apixaban,BMS-562247),a highly potent,selective,efficacious,and orally bioavailable inhibitor of blood coagulation factor Xa. 2007. DOI: org/10.1021/jm070245n.
5. Discovery of potent, efficacious, and orallybioavailable inhibitors of blood coagulation factor Xa with neutral P1 moieties. DOI: 10.1016/j.bmcl.2006.08.027.
6. 阿哌沙班创制的多维优化. 郭宗儒
7. 凝血因子Xa直接抑制剂的研究现状及应用. 2018.