替普瑞酮是临床上常用的抗胃溃疡药物,近年来发现其同时也是一种无明显不良反应的HSP诱导剂,可诱导各种组织器官中HSP的表达并发挥多种保护作用。
一、替普瑞酮简介
替普瑞酮,又名戊四烯酮,是树汁中的萜类物质,黄色油状液体,可溶于乙醇等有机溶剂,油水分配系数约为9,难溶于水,是日本卫材制药株式会社于1984年上市的一种新型抗消化性胃溃疡药。近年来随着研究的深入,发现替普瑞酮可作为一种无明显不良反应的HSP诱导剂,其结构中的类维生素A骨架能够诱导各种组织器官,如胃黏膜、小肠、大脑、心脏、肝脏、肾脏、肺、视网膜中HSP的表达[8,9,10,11],许多研究发现在缺血、缺氧、氧化刺激等应激情况下,替普瑞酮通过增加体内HSP的表达对各种组织器官起保护作用[8,11,12,13]。因此,替普瑞酮在组织器官保护方面有良好的应用前景。2001年Ooie等[13]首次发现替普瑞酮能通过诱导心脏HSP72表达发挥抗心肌缺血再灌注损伤作用,这一发现引起了广泛关注,并在此基础上对替普瑞酮的心肌保护作用及其机制进行了更深入的研究。
二、替普瑞酮与心肌缺血再灌注损伤
1.诱导HSP70/HSP72表达:
在大鼠心脏缺血再灌注模型中,口服单剂量(200 mk/kg)的替普瑞酮能够通过诱导心脏HSP72表达发挥抗心肌缺血再灌注损伤的作用[13,14,15,16]。在复灌注期间,替普瑞酮预处理可明显减少缺血20或40 min模型中肌酸激酶的释放,并且左心室功能明显恢复;口服替普瑞酮24 h后HSP72表达达到高峰值,虽然HSP72的表达呈剂量依赖性,但口服单剂量达200 mg/kg时,诱导HSP72表达的药效达到[13]。因此,许多基础研究以200 mg/kg作为动物模型的给药剂量。目前尚无研究证实替普瑞酮同样可诱导人体心脏内HSP72表达,但是有研究证实口服替普瑞酮(600 mg)能明显增加人体外周血单核细胞HSP72及HSP90表达[17]。另外,胰岛素抵抗能减弱替普瑞酮对HSP72的诱导及其抗心肌缺血再灌注损伤作用[16](图1)。
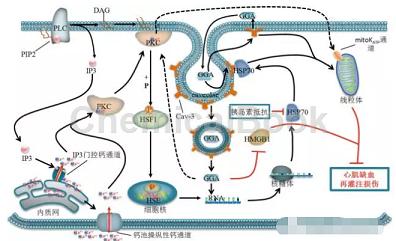
图1 替普瑞酮在心肌缺血再灌注损伤中的保护作用及其机制示意图(替普瑞酮可能参与了PKC信号转导系统的激活,使细胞内游离Ca2+浓度升高,从而通过激活PKC调节HSP70的表达,HSP70在心肌线粒体保护作用中发挥重要作用,并且替普瑞酮能诱导Cav-3表达上调,Cav-3介导对替普瑞酮的摄取及HSP70的胞膜窖定位,也是HSP70激活及随后线粒体呼吸之间的重要导线,因此线粒体是替普瑞酮心肌保护作用的靶点,同时PKC可能参与激活mitoKATP通道从而保护线粒体呼吸功能)
在后续研究中,许多研究者对替普瑞酮抗心肌缺血再灌注损伤作用机制进行了深入探索。Yamanaka等[14]研究发现蛋白激酶C(protein kinase C,PKC)在替普瑞酮诱导HSP72表达和心肌保护作用中具有至关重要的作用。替普瑞酮能够诱导PKC转位并激活,活化的PKC进一步使热休克因子1(heat shock factor 1,HSF1)磷酸化,随后磷酸化的HSF1从细胞质转位至细胞核内并且结合于HSP72基因的热休克元件(heat shock elements,HSE),进而启动HSP72基因表达并发挥心肌保护作用(图1)。但替普瑞酮诱导的PKC激活的信号通路仍有待进一步研究,其可能参与了PKC信号转导系统的激活,细胞内游离Ca2+浓度升高在此过程发挥重要作用(图1)。研究还发现替普瑞酮能通过抑制高迁移率族蛋白1(high mobility group box 1 protein,HMGB1)表达减轻缺血再灌注损伤[18](图1)。
此外,线粒体是替普瑞酮抗缺血再灌注损伤的作用靶点[15]。替普瑞酮能通过诱导HSP72表达,减轻心肌缺血再灌注损伤引起的线粒体超微结构的损害,改善心肌细胞线粒体呼吸,从而发挥抗心肌缺血再灌注损伤的作用(图1)。并且线粒体ATP敏感性钾通道(mitochondrial ATP-sensitive potassium channel,mitoKATP channel)的开放可能参与了替普瑞酮线粒体保护作用。Kitahata等[19]同样发现替普瑞酮的心肌保护作用可能与mitoKATP通道相关。而mitoKATP通道的激活受到PKC调控[20],因此替普瑞酮可能通过诱导PKC转位并激活mitoKATP通道进而保护线粒体功能,从而发挥抗心肌缺血再灌注损伤作用。
2.诱导窖蛋白3表达:
替普瑞酮的抗缺血再灌注损伤作用还与胞膜窖和窖蛋白3有关。替普瑞酮能通过诱导窖蛋白3的表达与胞膜窖的形成,从时间与空间上优化经线粒体保护性信号传导通路而发挥心肌保护作用[21]。窖蛋白3参与胞膜窖对替普瑞酮的摄取和介导HSP70的胞膜窖定位与激活,并且直接影响线粒体功能(图1)。此外,另一项研究发现联合应用替普瑞酮和异氟烷预处理能发挥协同抗心肌缺血再灌注损伤作用,并且这一保护作用同样受到窖蛋白3的介导[21]。Kitahata等[19]观察到七氟烷也能够强化替普瑞酮的此种心肌保护作用,并且该保护作用与诱导HSP70表达相关。但是这些研究均无法证实诱导HSP70表达和心肌保护作用之间存在直接联系。
3.保护内皮功能:
口服替普瑞酮能减轻缺血再灌注损伤导致的心脏血管内皮功能障碍而发挥心肌保护作用,PI3激酶及Rho激酶可能参与了该保护作用[22]。此外有研究发现替普瑞酮的内皮保护作用与诱导HSP90表达相关,其可激活HSP90-AMPK-eNOS-NO信号通路而改善内皮功能障碍[17]。可以看出替普瑞酮的抗缺血再灌注损伤作用与其诱HSP70/HSP72表达密切相关。在乳鼠心肌细胞建立缺氧复氧模型中,HSP72特异的小干扰RNA消除了替普瑞酮对HSP72的诱导表达作用并且替普瑞酮此种保护作用消失,因此体外实验证实替普瑞酮的心肌保护作用与HSP72存在直接联系[15]。但在许多体内实验中发现除了热休克蛋白,替普瑞酮还能诱导其他保护性蛋白的合成,如NOS、硫氧还蛋白等。所以HSP72尚不能作为替普瑞酮抗缺血再灌注损伤作用的唯一解释。有研究观察到在大鼠心肌缺血再灌注损伤模型中,替普瑞酮对HSP27、HSP60、NOS(包括iNOS、nNOS及eNOS)、硫氧还蛋白等保护性蛋白并没有诱导表达作用[13],其他保护性蛋白,如HSPB5、HSPB8、HSP90等的作用仍有待研究。虽然目前的体内实验只能提供一些排除性证据,但是仍然可以确定HSP72在替普瑞酮抗心肌缺血再灌注损伤作用中具有核心地位。
三、替普瑞酮与心房颤动(房颤)
1.抑制房颤的发生:
各种器质性心脏病的早期与加剧过程常伴随电重构,主要是心肌细胞动作电位时程(action potential duration,APD)发生适应性延长,使Ca2+内流代偿性增加,进而使心肌收缩力代偿性增强。但电重构在发挥代偿作用的同时也将引起细胞内钙超载和异常触发活动事件,成为各种心律失常发生的基质。许多研究证实诱导HSP表达能够阻止房颤的致心律失常基质产生[23]。作为HSP诱导剂,替普瑞酮能够改善各种原因,如心肌缺血、心力衰竭(心衰)等,引起的电重构改变,从而抑制房颤的发生。
替普瑞酮可减轻缺血引起的心房传导异常从而抑制缺血相关房颤的发生[24],这种保护作用表现为抑制缺血部位有效不应期(effective refractory period,ERP)延长、传导迟滞及传导模式的改变。该研究观察到替普瑞酮对犬心房缺血模型中HSP72(并非HSP27)的表达有明显诱导作用,其可能通过诱导HSP72表达在抑制缺血诱导房颤基质产生的作用中发挥重要作用。在兔的心衰模型中,替普瑞酮同样可通过诱导HSP表达抑制ERP与APD延长,可逆转心衰所致的电重构,减少触发活动及APD离散度从而降低房颤诱发率[25]。这些保护作用可能与其诱导各种钠通道及钾通道蛋白表达和维持钙稳态相关。其能够诱导Cav1.2、Nav1.5、Kir2.1、Kv1.4、Kv7.1和Kv11.1等离子通道蛋白与心肌细胞肌浆网钙ATP酶(sarcoplasmic reticulum Ca2+-ATPase,SERCA2a)mRNA的表达,从而缩短ERP,减弱心肌细胞对APD交替的易感性及抑制触发活动。
2.抑制房颤的进展:
心房重构所引起的电生理变化增加房颤启动和持续的可能性,其生理电重构主要表现为ERP和APD缩短。多项研究表明替普瑞酮可通过诱导HSP表达抑制房颤进展。在细胞房颤重构模型中,Brundel等[26]发现替普瑞酮可抑制节律加速细胞内L型钙电流的减弱及APD缩短,使钙瞬变及细胞收缩减弱,此种保护作用需要HSP27的表达及磷酸化。他们还通过快速心房起搏建立了实验犬房颤重构模型,发现替普瑞酮抑制了心动过速导致的ERP缩短及房颤的启动。Brundel等[27]还发现替普瑞酮几乎能够完全抑制房颤细胞模型中节律加速所致的肌纤维溶解,从而抑制房颤的进展。
以上2项研究均认为替普瑞酮抑制房颤进展的作用与HSP27而非HSP70相关,他们都通过基因转染发现HSP27过表达可复制替普瑞酮在实验中所观察到的保护作用。其中一项研究还发现通过敲除HSP27基因可阻断替普瑞酮介导的保护作用[26]。另一项研究还将结果扩展到了房颤患者,在阵发性及持续性房颤中,HSP27水平与房颤持续时间及肌溶解数量呈负相关[27]。体内实验中,Zhang等[28]建立了可行基因操作的果蝇房颤模型,发现替普瑞酮和DmHSP23过表达均可改善节律加速心肌细胞的重构及果蝇心脏收缩功能障碍,而该研究认为DmHSP23与人类HSP27在功能上属直系同源。因此,替普瑞酮可通过诱导HSP27表达抑制心房重构和房颤的进展。
尽管诸多实验均证实替普瑞酮对房颤有治疗作用,但是由于替普瑞酮的油水分配系数很高,大约为9,因此需要高剂量才能起效[24,26]。为了克服这一缺点,荷兰的HALT&REVERSE项目合成了替普瑞酮的各种衍生物,拥有更佳的药理化学特性及促HSP表达作用[29,30]。并且替普瑞酮及其衍生物的HSP诱导作用呈刺激依赖性,在正常情况下其HSP诱导作用较弱,预示着其不良反应较少[30]。其中一些衍生物在体外实验中被证实存在保护作用,并且其中一种衍生物将被应用于房颤动物模型。此外,已有相关研究在心脏手术后的患者中测试替普瑞酮能否诱导心房HSP表达及抑制术后房颤的发生[29]。
四、替普瑞酮与动脉粥样硬化
动脉粥样硬化是一种慢性炎症性疾病,是心血管疾病的主要病理基础。内皮细胞黏附分子(endothelial cell adhesion molecules,ECAM)介导的组织内异常白细胞聚集参与了动脉粥样硬化慢性炎症反应过程。替普瑞酮可增强高温(42 ℃)刺激对人体动脉内皮细胞HSP70的诱导表达作用,进一步阻止肿瘤坏死因子(TNF)α诱导的核因子(NF)-κB激活,进而减弱ECAM的表达,抑制内皮异常白细胞聚集[31]。此外,替普瑞酮还可通过抑制NF-κB激活减弱血管平滑肌细胞iNOS的表达,阻止炎症状态下NO的过量合成从而减轻细胞损伤及炎症反应,表明其可调节动脉粥样硬化等心血管疾病的病理生理过程[32]。胰岛素抵抗是动脉粥样硬化性心血管疾病发生、发展的危险因素之一。替普瑞酮可通过诱导HSP70增强衰老小鼠胰岛素敏感性从而延长寿命[33],研究还发现替普瑞酮在改善高脂饮食小鼠胰岛素抵抗的同时还能改善腹形肥胖,其机制与诱导HSP72表达及抑制氨基末端激酶(jun-NH2-terminal kinase, JNK)激活有关[34]。
五、替普瑞酮与心肌细胞凋亡
心肌细胞凋亡是物理化学等有害刺激,如湿热刺激、化疗药物引起心肌损伤的重要机制之一,在各种心血管疾病的病理过程中发挥重要作用。在小鼠的湿热刺激模型中,替普瑞酮能通过增加HSP70表达抑制湿热刺激诱导的心肌细胞凋亡,其机制主要是通过增加Bcl-2表达及抑制线粒体内细胞色素C释放从而抑制线粒体介导的凋亡[35]。最近的一项研究发现替普瑞酮还能通过诱导HSP70表达抑制应激诱导的心肌细胞凋亡,并且HSP70能通过竞争性与FAF1结合抑制Fas信号通路激活进而抑制凋亡[36]。此外,在小鼠的结蛋白相关的心肌病(desmin-related cardiomyopathy,DRM)模型中,替普瑞酮通过诱导HSPB8表达从而抑制线粒体细胞色素C释放、caspase-3激活和心肌细胞凋亡[37]。替普瑞酮在与化疗药物多柔比星联用时还能通过阻断RHO/ROCK1途径抑制多柔比星诱导的心肌细胞死亡、氧化应激损伤及心脏功能障碍而发挥心肌保护作用,同时还能增强多柔比星的抗肿瘤作用[38]。另有研究发现替普瑞酮具有明显的促凋亡作用,在很低浓度(0.1 μmol/L)时就能诱导白血病细胞HL-60凋亡[39]。
六、替普瑞酮的其他心肌保护作用
HSPB5基因Arg120Gly(R120G)位点错义突变会引起结蛋白相关的心肌病。研究发现替普瑞酮不仅能诱导R120G转基因小鼠体内HSPB8及HSPB1表达,还能抑制心脏扩大及间质纤维化,从而恢复心脏功能和促进存活;HSPB8过表达同样能抑制R120G小鼠心肌病的进展[37]。此外,Marunouchi等[40]发现替普瑞酮通过诱导HSPB1及HSPB8表达减轻心肌梗死后线粒体能量合成障碍而改善心脏收缩功能。因此,替普瑞酮也许能通过诱导sHSP表达成为治疗DRM及心肌梗死后心衰的新方法。
综上所述,作为一种无明显不良反应的HSP诱导剂,替普瑞酮可发挥多种心肌保护作用。替普瑞酮可改善心肌缺血再灌注损伤,抑制各种原因引起的房颤的发生与进展,改善动脉粥样硬化,减轻湿热刺激及化疗药物所致的心肌损伤,对DRM、心衰及胰岛素抵抗也有一定的治疗作用。此外,替普瑞酮还可以改善内皮功能,抑制心肌细胞凋亡、肌溶解及保护线粒体功能。值得注意的是,阿司匹林在心血管疾病患者中广泛应用,以往研究发现替普瑞酮对阿司匹林所致的胃黏膜损伤亦有一定保护作用[41],与质子泵抑制剂相比替普瑞酮是否可使心血管疾病患者获益更多?虽然其胃黏膜保护作用在临床上得到广泛认可,但其心肌保护作用还仅局限于动物实验,具体作用机制还不甚清楚。替普瑞酮也许可以为临床各种心血管疾病提供新的治疗方式,其各种衍生物具有更佳的药物化学特性及更强的HSP诱导作用,但是替普瑞酮的心肌保护作用还有待进一步证实。